Interaction et réseau de régulation
Préambule
Ce chapitre est dans la lignée du deuxième chapitre sur E. coli. Il a pour objectif de montrer aux non-biologistes comment les biologistes perçoivent une interaction ou un réseau. Le chapitre d'après visera le but opposé: montrer aux biologistes comment les mathématiciens (et/ou informaticiens) décrivent une interaction ou un réseau. Dans ce chapitre, j'ai volontairement fait l'effort d'aller relativement en profondeur dans certains paragraphes de manière à permettre aux biologistes d'y extraire une information plus concrète/spécifique. Il y a donc parfois deux niveaux de lecture et certains paragraphes sembleront un peu lourds à un lecteur avide de généralités ou de clarté. Selon moi, la structure/le plan et le message global de ce chapitre comptent plus que les détails. Si ces derniers vous gênent: n'ayez pas peur de lire en diagonale.
Interaction et réseau de régulation
Dans le chapitre précédent, nous avons dressé un portrait statique du fonctionnement de la bactérie E. coli. Nous allons maintenant décrire ce qui permet de "dynamiser" la bactérie. Cette dernière n'est pas un simple sac de molécules indépendantes. Les bactéries contrôlent et ajustent en permanence le niveau d'expression de chacun de leurs gènes pour adapter le contenu cellulaire aux conditions extérieures. Ce contrôle de l'expression des gènes est effectué grâce à un réseau de régulation génique. Un réseau de régulation génique est une collection de gènes qui interagissent les uns avec les autres via leurs produits d'expression (les protéines), permettant un contrôle mutuel de leurs taux d'expression. Les réseaux de régulation génique sont, avec la régulation du métabolisme, les principaux déterminants de l'adaptation d'une bactérie à son environnement.
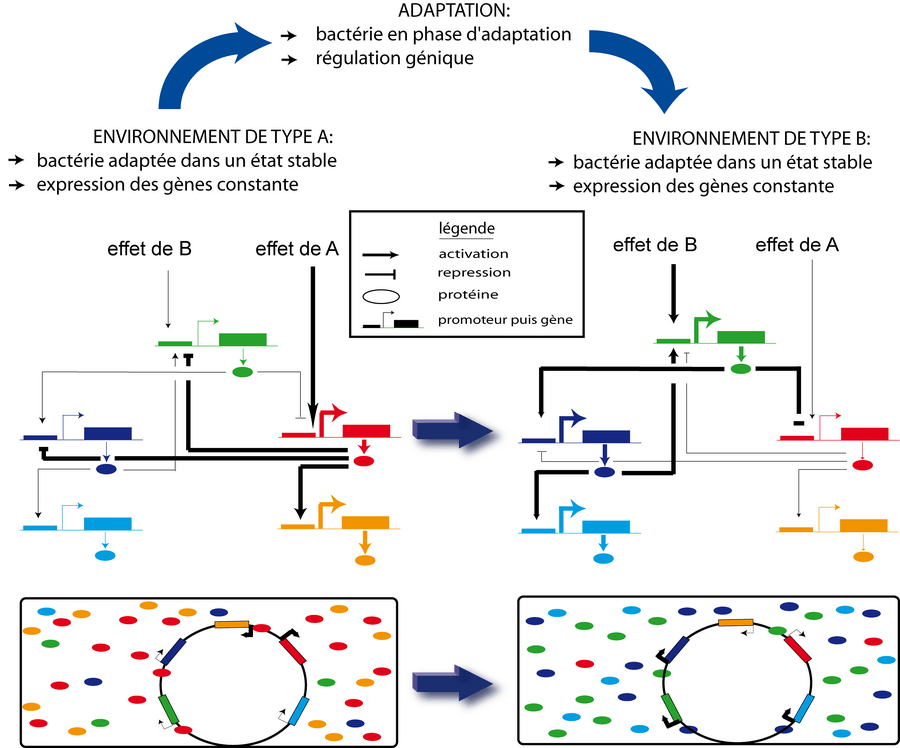
Un réseau peut induire une fonction que ses sous parties ne possèdent pas: le fameux truisme " le tout est plus que la somme des parties". On qualifie ce phénomène d'émergence. La vie (d'une bactérie) émerge de l'ensemble des interactions des molécules internes à la bactérie. Ces interactions forment le réseau de régulation. Notons que la notion d'émergence fait l'objet d'une polémique en philosophie des sciences. Loin d'être un fait établi par la méthode scientifique, l'émergence ne serait qu'un simple mot utile pour décrire des phénomènes complexes que l'on ne comprend pas.
Un réseau est un ensemble de noeuds reliés entre eux par des liens (ou des flèches dans le cas d'un graphe orienté). Dans un réseau de régulation, les noeuds sont des molécules et les liens représentent une interaction physique entre deux molécules. Contrairement à un circuit intégré, au réseau neuronal ou au réseau internet qui possèdent des liens bien réels, les flèches d'un réseau de régulation sont virtuelles: elles reposent sur les lois de la diffusion. Ces lois introduisent des propriétés probabilistes au réseau de régulation. Un composé A qui baigne dans le cytoplasme d'une bactérie se déplace de manière aléatoire (le mouvement brownien) et a donc une certaine probabilité de rencontrer un composé B en un temps t. Si la rencontre entre les composés A et B induit un changement/effet/modification/conséquence, alors il existe un lien entre A et B.
- Si l'enzyme A (par exemple la phosphoglucose isomerase) rencontre le métabolite B (le glucose-6-phosphate), le changement correspond à la disparition du substrat (le glucose-6-phosphate) et l'apparition du produit (le fructose-6-phosphate). On parle, pour ce type d'interaction, de réaction métabolique.
- Si l'enzyme A (par exemple UhpB) rencontre une protéine B (UhpA), le changement correspond à la modification chimique de la protéine B (la phosphorylation d'UhpA). On parle, pour ce type d'interaction, de "transduction du signal" ou de "modification post-traductionnelle".
- Si le facteur de transcription A (par exemple LacI) rencontre la séquence promotrice du gène B (le gène lacZ), le changement correspond à une modification de l'expression du gène B (répression du gène lacZ). On parle, pour ce type d'interaction, de régulation génique/ régulation de l'expression génique /régulation transcriptionnelle.
Cette liste n'est pas exhaustive et on compte d'autres types d'interactions comme, par exemple, les interactions mettant en jeu des ARN ou des interactions intercellulaires.
On compte actuellement plus de 1326 réactions métaboliques chez E. coli. Ces voies métaboliques sont en général assez linéaires (glycolyse, cycle de Krebs) et ressemblent assez peu à un réseau: la connectivité est faible: Il y a peu de boucles de rétroaction. Le temps d'une réaction enzymatique est inferieur à la seconde. L'ajustement des pools de métabolites et des flux est donc très rapide et se fait également à l'échelle de la seconde.
La transduction du signal est assez peu décrite chez E. coli. On compte cependant une trentaine de systèmes de phosphorylation dits à deux composantes. Il existe aussi un système de transfert de phosphate (PTS) chargé de faire rentrer le glucose dans la cellule. La transduction du signal semble aussi être assez linéaire. Ces modifications chimiques, parfois sous forme de cascade, sont très rapides et s'effectuent, comme les réactions métaboliques, à l'échelle de la seconde.
On compte pour l'instant plus de 4792 régulations transcriptionnelles. Ces régulations forment un réseau avec de nombreuses boucles de rétroactions. Il est ainsi difficile de savoir intuitivement comment évolue un tel système. Ces régulations se font plutôt à l'échelle des minutes, voire des heures. En effet, si la fixation du facteur de transcription au promoteur prend quelques secondes, l'activation ou la répression de la transcription du gène cible ne montrera ses effets que lorsque la protéine cible résultante se sera accumulée suffisamment ou lorsqu'elle se sera dégradée suffisamment.
Cliquer pour voir le code Matlab
% Ce petit script permet d'importer la strcuture d'une protéine et de la
% visualiser. Si il ne fonctionne pas, verifier votre connection internet
%ainsi que votre proxy (à parametrer dans matlab)
ubi = getpdb('1ZRC');% recupere la structure 3D
%de Crp en complexe à l'ADN dans Protein Data Bank (PDB) database
h1 = molviewer(ubi);% affiche la molecule
evalrasmolscript(h1,['background white; spin; cartoons on'...
';wireframe off;cpk off;moveto 15 1 0 0 90']);
% paramètre pour un affichage adequate (codé en Rasmol)
Dans cette thèse, je parlerai de réseaux de régulation globaux lorsque j'intégrerai tous les mécanismes de régulation présents dans une cellule pour lui permettre de s'adapter. A l'inverse, je parlerai de réseau de régulation géniques lorsque nous étudierons des réseaux construits à partir de régulations transcriptionnelles uniquement. La littérature s'intéresse principalement aux réseaux de régulation génique car c'est dans ce type de réseaux que l'on retrouve le plus de systèmes non linéaires. Cependant cette dichotomie est parfaitement illusoire tant métabolisme, régulations transcriptionnelles, transduction du signal et autres régulations (communication intercellulaire, ARN...) fonctionnent ensemble et s'influencent mutuellement. Le réseau de régulation global de la bactérie E. coli est un réseau extrêmement complexe dans sa topologie avec plusieurs dizaines de milliers/millions/milliards (?) d'interactions et de nombreuses boucles de rétroaction. On est encore loin d'avoir découvert toute la pluralité et la diversité des mécanismes de régulation. Ces mécanismes agissent de concert à des échelles de temps très différentes. Enfin, la nature intrinsèquement stochastique des mécanismes de régulation (dû au mouvement brownien) font qu'un réseau de régulation est idiosyncratique: j'entends par là le fait que la réponse d'un même réseau à une perturbation donnée peut être différente entre deux clones. Autrement dit, pour un même génotype, deux bactéries peuvent adopter deux phénotypes différents en réponse à une même perturbation. Nous reviendrons largement sur cette notion importante dans le chapitre sur le bruit.
Deux exemples clés
Je vais décrire le fonctionnement de deux réseaux de régulation parmi les plus connus et donc les mieux décrits à ce jour. Ces deux réseaux fonctionnent à des échelles de temps très différentes: le premier, la répression catabolique, fonctionne à l'échelle de la dizaine de minutes alors que le deuxième, le chimiotactisme fonctionne à l'échelle de la seconde. Pour ces deux exemples, je suis volontairement rentré dans les détails. Ce niveau de détail enchantera celui qui veut comprendre ces systèmes en profondeur c'est-à-dire celui qui s'apprête probablement à les utiliser. Cependant, si vous souhaitez uniquement avoir une idée générale de la manière dont les biologistes décrivent ces réseaux, alors contentez vous de lire les paragraphes qui suivent de manière superficielle. Essayez juste de capter la "philosophie" sous-jacente en vous concentrant sur le champ lexical utilisé pour décrire les interactions.
La répression catabolique
Pourquoi la bactérie E. coli a t'elle besoin d'un réseau de régulation pour s'adapter? Pourquoi, par exemple, ne pas produire chacune des 4500 protéines en 100 exemplaires pour être sûr de répondre à n'importe quel imprévu ? Par exemple, être prêt à manger immédiatement n'importe quel sucre qui se présenterait dans le milieu extérieur ?
Maintenir des protéines inutiles dans la cellule a un coût. Ce dernier impacte directement le fitness de la bactérie, c'est-à-dire sa capacité à se reproduire. Une bactérie, qui au fil de l'évolution, acquiert la capacité de ne synthétiser une protéine que lorsqu'elle en a vraiment besoin, augmentera son fitness et sera ainsi sélectionnée par l'évolution.
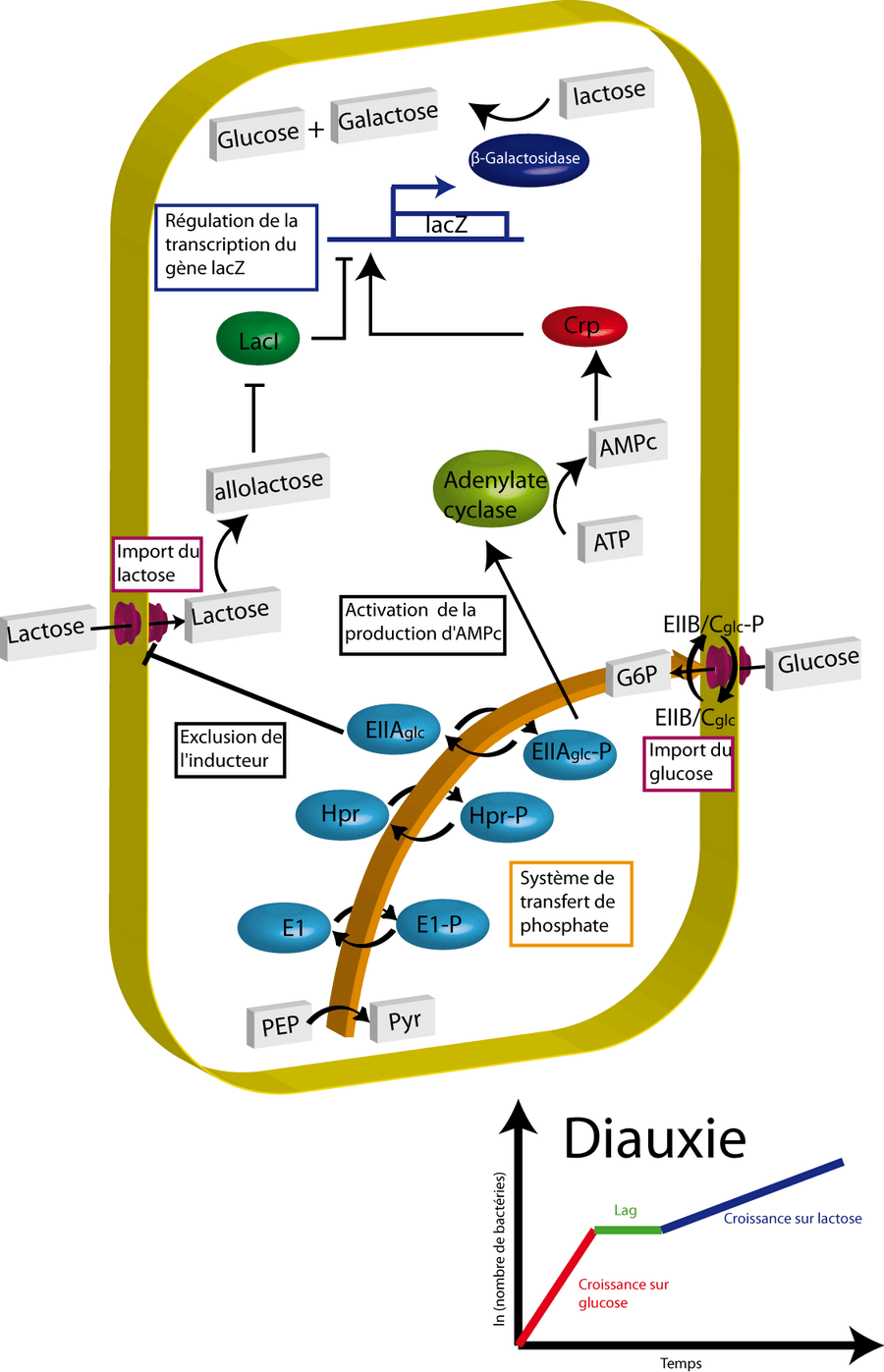
Je vais maintenant décrire le premier mécanisme de régulation génique découvert: la régulation de l'opéron lac. Dans son environnement naturel, l'opéron lac code pour des protéines nécessaires à la digestion du lactose. La cellule peut utiliser le lactose comme source d'énergie en produisant une enzyme la β-galactosidase (codée par le gène lacZ). Cependant, il est inutile de produire cette enzyme s'il n'y a pas de lactose dans le milieu ou si une source de carbone plus facilement métabolisable comme le glucose est présente dans le milieu. L'expression de l'opéron lac est contrôlée par plusieurs mécanismes qui assurent que les dépenses d'énergie pour produire la β-galactosidase n'ont lieu que lorsque c'est nécessaire. Ces mécanismes de régulation ont pour conséquence l'utilisation séquentielle du glucose puis du lactose en deux phases distinctes de croissances. On appelle cela une diauxie (Monod 1942).
Le premier mécanisme de contrôle est la réponse régulatrice due à la présence de lactose dans le milieu. Ce mécanisme utilise une protéine régulatrice intracellulaire (un facteur de transcription) nommée répresseur LacI qui bloque la transcription du gène lacZ et donc la production de β-galactosidase. Si le lactose est absent du milieu de culture, ce répresseur se fixe fermement à une courte séquence d'ADN appelée l'operateur lacO juste en amont de l'origine du gène lacZ. La fixation du répresseur à son site operateur interfère avec la fixation de l'ARN polymérase au promoteur et empêche ainsi la transcription de l'opéron. Quand la cellule pousse en présence de lactose, un métabolite issu du lactose appelé allolactose se fixe au répresseur LacI induisant un changement de sa conformation. Ainsi altéré, le répresseur est incapable de se fixer à l'operateur lacO et permet à l'ARN polymérase de transcrire le gène lacZ. Ainsi la concentration de β-galactosidase augmente fortement et le lactose peut être métabolisé.
Le second mécanisme de contrôle est la réponse régulatrice due à l'absence de glucose dans le milieu. Ce mécanisme utilise le régulateur global Crp pour augmenter la production de β-galactosidase en absence de glucose. L'AMPc est une molécule dont la concentration dans la cellule est inversement proportionnelle à celle du glucose. Cette molécule fixe Crp formant ainsi le complexe Crp--AMPc (Kolb et al. 1993). Ce complexe peut alors se fixer au site de liaison de Crp situé dans la région promotrice en amont de l'origine du gène lacZ puis aider au recrutement de l'ARN polymérase. En absence de glucose, la production d'AMPc est forte et le complexe Crp--AMPc se fixe à l'ADN augmentant très fortement la production de β-galactosidase. A l'inverse, en présence de glucose, la concentration d'AMPc est faible, la formation du complexe n'a pas lieu et Crp ne peut pas se lier à l'ADN diminuant drastiquement la transcription de l'opéron lac et donc la production de β-galactosidase. Mais comment le glucose régule-t'il la concentration d'AMPc et donc la transcription de l'opéron lac ? L'import de glucose est medié par le système de transfert de phosphate (PTS) (Postma et al. 1993). Ce système transfère séquentiellement un groupement phosphate du Phosphoenolpyruvate (PEP) jusqu'au glucose formant ainsi du glucose-6-phosphate. La séquence de transfert des phosphates fait intervenir 4 enzymes dont l'enzyme EIIAglc. Lors de l'import du glucose, cette enzyme est sous forme déphosphorylée or seule la forme phosphorylée est capable de fixer puis d'activer l'adenylate cyclase, l'enzyme responsable de la biosynthèse de l'AMPc.
Le dernier mécanisme de contrôle est appelé mécanisme de l'exclusion de l'inducteur (Saier and Crasnier 1996). Lorsque le glucose est importé, l'enzyme EIIAglc est sous forme déphosphorylée. Sous cette forme, l'enzyme est capable de se fixer au transporteur du lactose et d'inactiver ainsi son transport. L'entrée de l'inducteur est ainsi empêchée. Or ce dernier est chargé d'inactiver le répresseur LacI. En conséquence, en présence de glucose, le répresseur reste sous sa forme active et réprime la transcription de l'opéron lacZ.
J'ai choisi de développer cet exemple de la répression catabolique pour plusieurs raisons. Premièrement car le haut niveau de détail dans la compréhension des mécanismes rend son étude intéressante pour les modélisateurs (Kremling et al. 2009). Deuxièmement, car il illustre parfaitement les liens très étroits qui existent entre régulation transcriptionnelle, transduction du signal et métabolisme. Troisièmement car une des publications présente dans cette thèse remet en cause le modèle actuel de répression catabolique que je viens de vous décrire.
Le chimiotactisme
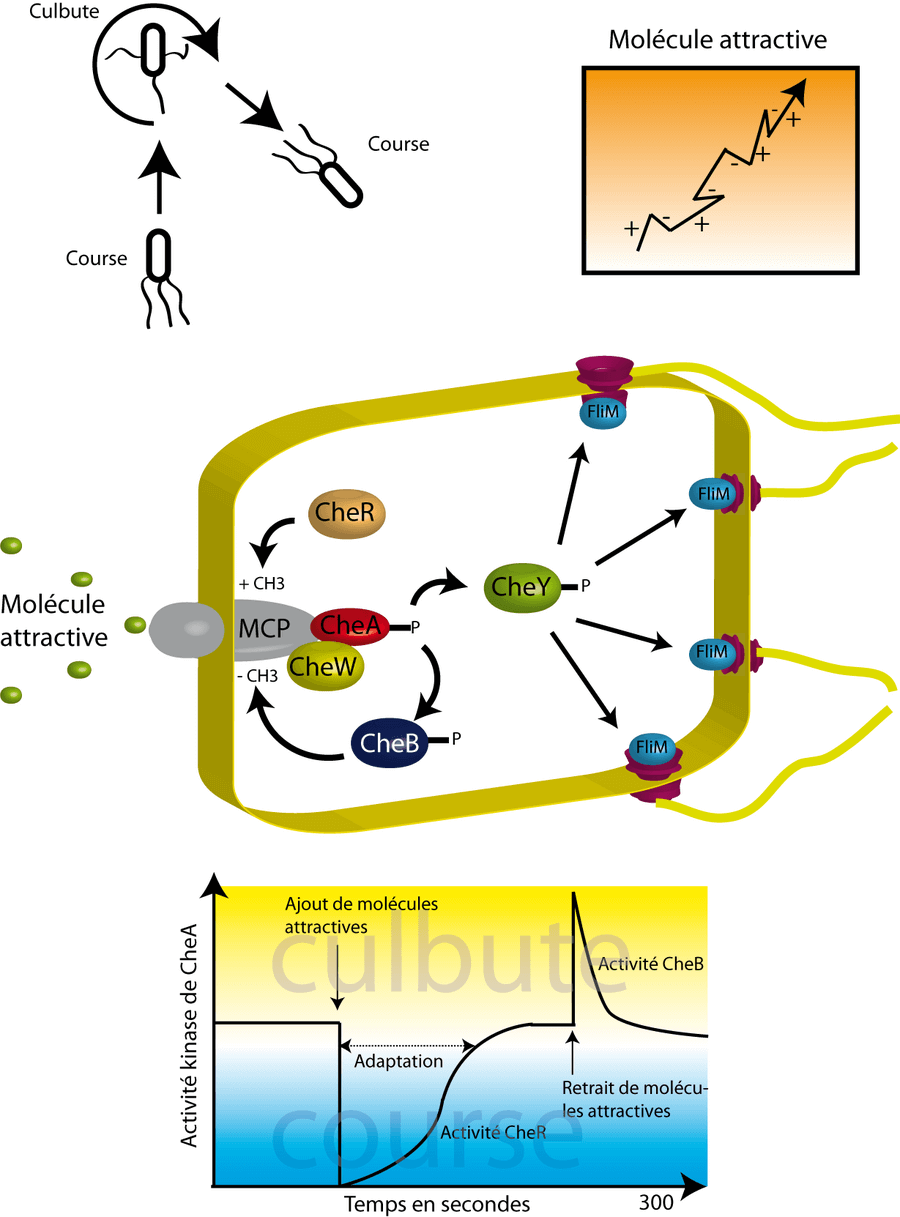
Le chimiotactisme est défini comme un mouvement orienté vers ou dans le sens opposé d'une substance chimique. Les bactéries E. coli, capables de se mouvoir grâce à leurs flagelles, sont attirées en direction de certains nutriments (sucres, acide aminés) ou à l'inverse repoussées par des substances toxiques (alcool, acide gras).
Pour réussir à modifier leur direction vers une substance attractive, les bactéries doivent forcement mesurer le gradient de concentration. Cependant la différence de concentration d'une substance attractive entre l'avant et l'arrière de la cellule est trop faible (contrairement aux cellules eucaryotes) pour être détecté. Or, si la mesure du gradient n'est pas spatiale alors c'est qu'elle est temporelle. Autrement dit, cela signifie que les bactéries sont capables de "mémoriser" les concentrations. Petit bémol important que je préfère ajouter quand j'utilise des analogies avec le système nerveux: la bactérie ne "choisit" pas son chemin et n'a pas "conscience" de se diriger vers un nutriment.
La motilité des bactéries est permise par la présence des flagelles qui peuvent tourner dans deux sens: La rotation dans le sens inverse des aiguilles d'une montre arrange les flagelles en un seul faisceau synchrone; il en résulte la natation rectiligne de la bactérie. On appellera cette phase la course. Au contraire, lorsque la rotation se fait dans le sens des aiguilles d'une montre, les flagelles se désolidarisent les uns des autres, ainsi la bactérie bouge de manière erratique et tourne dans tous les sens sans vraiment avancer. On appellera cette phase la culbute.
Dans un environnement chimique isotrope, E. coli se déplace en "marche aléatoire" c'est-à-dire en alternant des épisodes de course et des épisodes de culbute. Lorsque la bactérie avance dans la bonne direction, la concentration de la substance attractive augmente. La bactérie détecte cette augmentation et diminue la probabilité de culbute. Les épisodes de course dans la bonne direction sont alors plus longs et cette marche aléatoire biaisée permet aux bactéries de migrer vers la substance attractive.
Le gradient chimique est perçu à l'aide d'un chémorécepteur transmembranaire appartenant à la famille des "methyl accepting chemotaxis proteins (MCPs)". Lorsque une substance attractive se fixe au récepteur, un signal est transmis à la partie cytoplasmique du récepteur où les protéines CheW et CheA sont fixées. Ce signal inhibe l'autophosphorylation de l'histidine kinase CheA. En absence de substance attractive, le groupe phosphoryl, résultant de l'autophosphorylation de CheA, est rapidement transféré aux régulateurs de réponse, CheB et CheY. CheY diffuse alors vers le moteur du flagelle et interagit avec la protéine FliM ce qui induit le changement du sens de rotation des flagelles: du sens inverse des aiguilles d'une montre au sens des aiguilles d'une montre. Ainsi l'absence de substance attractive induit la "culbute". A l'inverse, sa présence inhibe l'autophosphorylation de cheA, diminue la probabilité de "culbute" et permet ainsi une "course" plus longue.
La bactérie garde une sensibilité pour certaines substances attractives du nanomolaire jusqu'au millimolaire. En effet, il faut qu'elle soit capable de "traverser" jusqu'à 6 ordres de grandeur. Or, cette cascade de phosphorylation n'explique pas comment la bactérie se "réadapte" à la nouvelle concentration pour être à nouveau "attirable" par la concentration du dessus. Ce phénomène nommé adaptation compense les variations de concentration de la substance attractive en ajustant en permanence le niveau de methylation du récepteur. L'enzyme CheR ajoute jusqu'à 4 groupes méthyle sur le récepteur de façon constitutive. Plus de groupes méthyle sont rajoutés au récepteur, plus ce dernier active l'autophosphorylation de CheA. A l'inverse, une fois phosphorylé grâce à CheA, la protéine CheB agit comme une méthyle estérase et enlève des groupes méthyles sur la partie cytoplasmique du récepteur. Cette boucle de rétroaction négative permet d'ajuster en permanence le niveau de phosphorylation de CheA (et in fine le processus de décision de culbute) quelque soit la concentration de substance attractive présente. Ainsi, la bactérie reste sensible aux changements faibles, même pour les concentrations extrêmes. Cette régulation agit comme une "mémoire" [Petite parenthèse: Vous trouverez sans doute l'explication un peu difficile à comprendre. C'est peut être lié à un manque de clarté de ma part mais aussi lié au fait que le mécanisme de cet effet mémoire est difficile à se représenter cognitivement. Fin de la parenthèse] permettant à la bactérie de "se souvenir" des concentrations récentes et de les comparer avec les concentrations actuelles, donc de "savoir" si elle se déplace dans la bonne direction (Vladimirov and Sourjik 2009). Concluons sur le fait que tout comme la répression catabolique, ce mécanisme de chimiotactisme inspire également de nombreux travaux de modélisation (Bray et al. 2007).
La détection des interactions géniques
Deux bases de données Ecocyc (Keseler et al. 2009) et RegulonDB (Gama-Castro et al. 2008) référencent les interactions géniques décrites chez E. coli et indiquent la ou les méthodes qui ont permis de les détecter. Ces méthodes sont nombreuses et je vais prendre le temps d'en décrire quelques unes. Le choix de la méthode dépend de ce que l'on souhaite faire:
- Les méthodes in vitro permettent de démontrer l'existence d'une liaison physique ADN--protéine.
- Les techniques à haut-débit (Puce à ADN, ChIP-chip, RNA-Seq, chIP-seq) permettent de détecter in vivo un grand nombre d'interactions en même temps.
- Les gènes rapporteurs permettent de quantifier et contrôler l'effet d'une interaction in vivo et en temps réel.
- Les méthodes bioinformatiques essaient de se passer de l'expérimentation souvent longue et fastidieuse.
Rappelez-vous que les interactions les plus fiables sont évidemment celles qui sont confirmées par plusieurs méthodes et par plusieurs équipes. Si vous faite de la biologie synthétique, ne faites jamais confiance a priori à une interaction décrite dans la littérature. Vérifiez-la par vous-même et préférez l'utilisation des interactions (ou des briques) les plus classiques qui ont fait leurs preuves depuis longtemps (LacI, TetR, AraC etc...).
Les méthodes in vitro
Le retard sur gel
Le retard sur gel (EMSA ou electrophoretic mobility shift assay) est une technique de biologie moléculaire permettant de détecter une interaction entre une protéine et de l'ADN. Le principe consiste à faire migrer dans un gel d'électrophorèse, le fragment d'ADN d'intérêt sur une piste, et sur une autre piste, le même fragment d'ADN mis en contact avec la protéine susceptible de se fixer dessus. La piste contrôle devrait contenir une seule bande correspondant à l'ADN non lié. Si la protéine (le potentiel facteur de transcription) est capable de se fixer au fragment d'ADN, la migration de ce dernier sera plus lente (il sera retardé) et la bande correspondante sera décalée vers le haut. A l'inverse, si la protéine n'a aucune affinité pour le fragment d'ADN, les deux bandes apparaitront au même niveau sur le gel.
L'empreinte à la Dnase
L'empreinte à la Dnase est une technique de biologie moléculaire qui détecte les interactions protéine--ADN en utilisant le fait qu'une protéine liée à l'ADN protège souvent cette ADN d'un clivage enzymatique. Par exemple, le fragment d'ADN d'intérêt peut être amplifié par PCR en utilisant un oligonucleotide marqué en 5' au 32P. Il en résulte une molécule d'ADN double brin marquée radioactivement à une seule de ces extrémités. Le clivage par la Dnase va produire des fragments de toutes les tailles. Les fragments plus petits migreront plus loin que les fragments longs. Le gel est ensuite utilisé pour exposer un film photographique spécial. Le pattern de clivage de l'ADN en absence de protéine est comparé au pattern de clivage de l'ADN en présence de la protéine susceptible de lier l'ADN. Si cette dernière se lie à l'ADN, le site de liaison est protégé de la digestion enzymatique. Cette protection crée une zone claire sur le gel correspondant à "l'empreinte" de la protéine.
Les méthodes à haut-debit
Les puces à ADN
L'apparition des puces à ADN a engendré des centaines d'expériences d'analyse du transcriptome qui mesurent le niveau d'ARNm à l'échelle du chromosome entier. Ces analyses ont été très utilisées pour trouver les cibles des facteurs de transcription en identifiant les gènes dont l'expression change lorsque le gène codant pour un facteur de transcription est déleté. Cependant cette approche n'a eu qu'un succès limité. En effet, le réseau de régulation génique d'E. coli est extrêmement complexe et de nombreux facteurs de transcription régulent l'expression d'autres facteurs de transcription. Ainsi la délétion d'un facteur de transcription a souvent de nombreux effets secondaires sur l'expression des gènes. Si la transcriptomic est utile pour étudier les patterns globaux de transcription, il est beaucoup plus difficile d'identifier les cibles directes (le "régulon") d'un facteur de transcription donné.
La technique du ChIP-chip
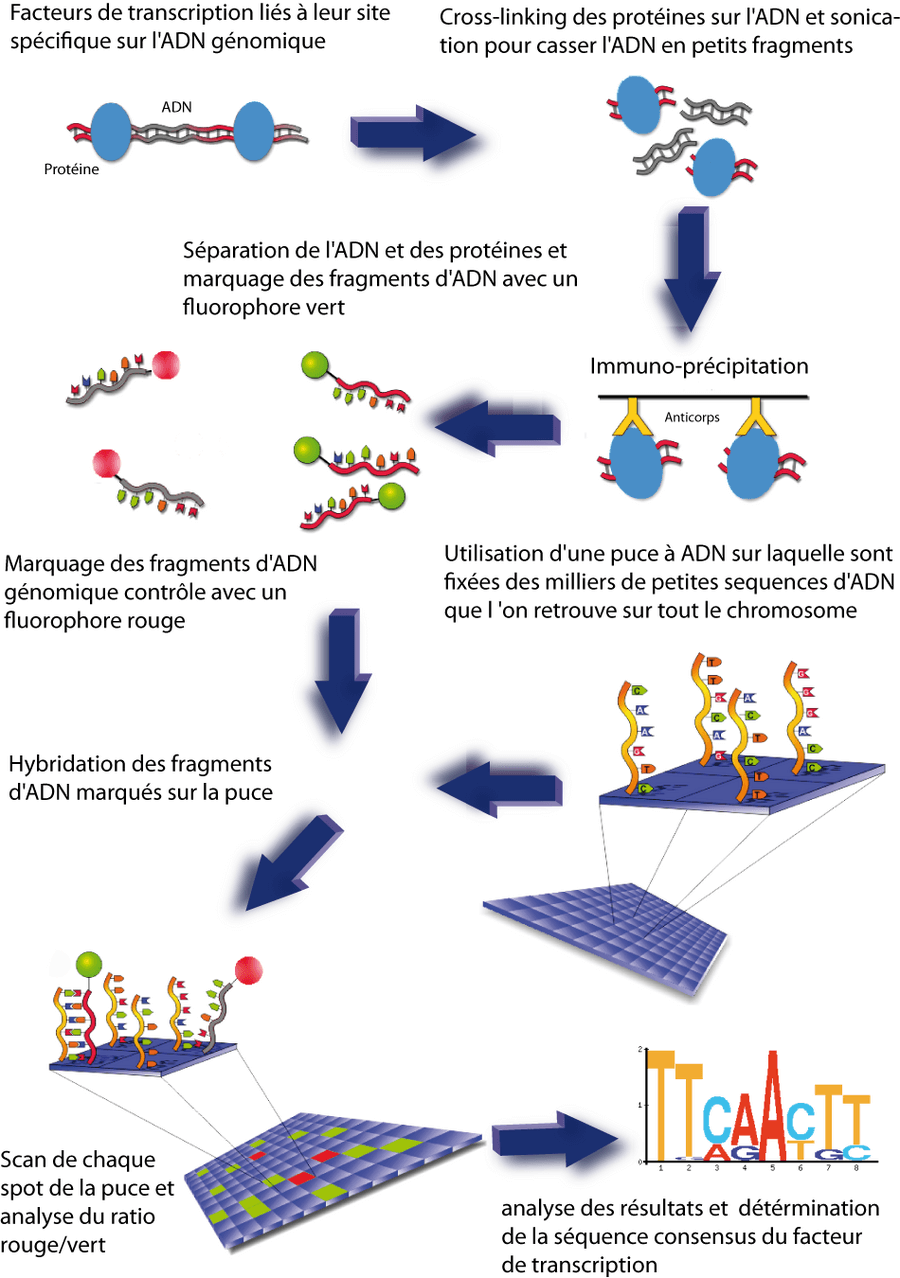
La méthode de Chromatin ImmunoPrecipitation on Chip (abrégée ChIP on chip) est une technique permettant de mesurer directement des interactions protéine--ADN in vivo. Il s'agit d'une combinaison de la technique de Chromatin Immunoprécipitation avec la méthode des puces à ADN. Contrairement à la transcriptomic, la mesure de cette interaction ne dépend pas de la transcription du gène en aval. Chez les bactéries, cette méthode est particulièrement adaptée pour déterminer, de manière globale, les sites de fixation sur le chromosome d'un facteur de transcription donné (Grainger and Busby 2008). Il est ensuite possible de déterminer la séquence d'ADN consensus de ce facteur de transcription.
Dans une expérience de ChIP-chip, les cellules sont fixées avec du formaldehyde ce qui crée des liaisons covalentes in vivo entre les facteurs de transcription et leur site de liaison sur l'ADN. Puis l'ADN est extrait des cellules et coupée en courts brins de l'ordre de 500pb par sonication. La protéine d'intérêt (associée aux fragments d'intérêt) est alors immuno-précipitée à l'aide de l'anticorps correspondant. On sépare ensuite l'ADN des protéines puis on marque les fragments d'ADN avec un fluorochrome vert. Puis on hybride ces fragments sur une puce à ADN avec un échantillon contrôle marqué, lui, en rouge. Ce contrôle, dont les données serviront de bruit de fond, peut être l'ADN génomique "faussement" immuno-précipité d'une souche délétée du facteur de transcription étudié. La puce est une plaque de petite taille sur laquelle sont fixés plusieurs dizaines de milliers de fragment d'ADN simple brin de 60 nucléotides. La séquence des ces milliers de fragments est espacée à intervalle régulier tout au long de la séquence du chromosome de E. coli. Ainsi les fragments immuno-precipités de plus de 500pb trouvent forcement leurs brins complémentaires auxquels s'hybrider. La plaque est ensuite lavée par des bains spécifiques pour éliminer les brins d'ADN ne s'étant pas hybridés. Chaque point (ou spot) de la puce va être analysée individuellement par un scanner à très haute résolution, et ce à la longueur d'onde d'excitation des fluorochromes rouges et verts. Puis, pour chaque spot, on détermine le ratio des intensités obtenues avec les deux fluorochromes. Les données finales correspondent à la valeur de ce ratio en fonction de la position sur le chromosome d'E. coli. Un ratio fluorochromes vert/rouge élevé signifie que l'hybridation de l'ADN immuno-précipité est supérieur à celle du contrôle ce qui signale la présence d'un site de liaison potentiel à cet endroit du chromosome.
Le RNA-Seq et le ChIP-Seq
Ces deux techniques récentes sont dérivées des deux précédentes mais cette fois la puce à ADN est remplacée par un séquençage à haut-débit. Comme dans l'analyse du transcriptome avec une puce à ADN, dans la technique de RNA-Seq, on s'intéresse au pattern global d'expression des gènes d'une souche donnée. Il peut s'agir d'un mutant d'un facteur de transcription donné que l'on compare avec une souche sauvage. Les différences d'expression entre les deux souches reflètent les effets de la délétion et donc les cibles potentielles du facteur de transcription. La première étape consiste à récupérer les ARN de la souche. Puis, si possible, on se débarrasse des ARN ribosomiaux. Les ARNm sont retrotranscris en ADNc puis fragmentés. Des séquences adaptatrices sont ajoutées à chaque fragment ADNc pour faciliter le séquençage. Chaque molécule est ensuite séquencée en utilisant les nouvelles technologies de séquençage à haut débit. En utilisant les données collectées, on peut créer une carte de transcription à l'échelle du génome qui indique le niveau d'expression de chaque gène/nucléotide (Wang et al. 2009). L'avantage énorme du RNA-Seq est son faible bruit de fond et sa grande résolution (1 nucléotide) supérieur à la résolution obtenue avec une puce à ADN (de l'ordre de la dizaine de nucléotides). Le chIP-Seq est similaire au ChIP-chip mais la puce à ADN est remplacée par le séquençage à haut-débit des ADN immunoprécipités.
Les techniques utilisant la biologie moléculaire
En biologie synthétique, l'expérimentateur ne souhaite pas uniquement avoir connaissance d'une interaction: il veut aussi l'utiliser, la contrôler. Pour cela, il doit utiliser des méthodes qui lui permettent de contrôler une interaction in vivo et en temps réel (pour ajuster la force de l'interaction pendant l'expérience). Pour cela, le recours à la biologie moléculaire devient indispensable: il faut manipuler, couper, coller, recombiner des séquences d'ADN. En théorie, la biologie moléculaire est facile mais en pratique elle est souvent difficile, longue et fastidieuse à cause de la présence de faux positifs que la théorie ne considère pas vraiment. Selon les personnes, le temps pour réaliser une construction de biologie moléculaire peut prendre entre quelques jours à plusieurs mois. Le plus frustrant en biologie moléculaire, c'est que l'acharnement nécessaire ne rapporte pas un résultat: il rapporte juste un outil, non publiable et qui ne fonctionnera peut être pas lorsque l'expérience l'utilisant sera réalisée. En résumé, un biologiste qui maitrise la biologie moléculaire sur le bout des doigts est un atout précieux dans un laboratoire ou une entreprise...
Les fusions transcriptionnelles
L'utilisation d'une fusion transcriptionnelle est une approche classique pour mesurer l'activité d'un promoteur dans une cellule vivante. Cette approche consiste à fusionner un promoteur d'intérêt en amont d'un gène rapporteur. Comme leurs noms l'indiquent, les gènes rapporteurs "rapportent" l'activité d'un promoteur.
Le premier gène rapporteur utilisé est le gène lacZ. Comme nous l'avons dit plus haut, ce gène code pour une enzyme, la β-galactosidase. Or la concentration de cette enzyme peut être facilement quantifiée en mesurant l'hydrolyse d'un substrat chromogenique (ONPG) (Miller 1972). L'apparition d'une couleur jaune (quantifiable à 420nm avec un spectrophotomètre) reflète la concentration de la β-galactosidase et donc l'activité du promoteur qui contrôle la transcription du gène lacZ. Cependant, la mesure de la concentration de β-galactosidase nécessite de lyser les cellules ce qui limite la résolution temporelle. De nouveaux gènes rapporteurs sont devenus de plus en plus utilisés et permettent de mesurer l'activité d'un promoteur in vivo (et sans avoir à tuer les bactéries), en temps réel et avec une très haute résolution temporelle. La Green Fluorescent Protein (GFP), dont la découverte a fait l'objet d'un prix Nobel en 2008 (Shimomura et al. 1962) émet une lumière verte (509nm) lorsqu'elle est excitée avec une lumière bleue (488nm). En quantifiant les photons émis à l'aide d'un photomultiplicateur, on peut mesurer la concentration de la GFP et remonter ainsi à l'activité du promoteur. A titre de parenthèse, lorsque le promoteur en amont du gène codant pour la GFP est très fort, on peut observer, à l'oeil nu, des colonies d'E. coli "vertes". Le deuxième rapporteur, la luciférase génère de la bioluminescence et émet donc des photons directement sans nécessité d'excitation. Comme pour la GFP, on peut mesurer l'activité d'un promoteur simplement en mesurant le nombre de photons émis par la luciférase. Dans le noir, après un petit temps d'adaptation, on peut observer, à l'oeil nu, la bioluminescence émise par des colonies d'E. coli.
Les fusions transcriptionnelles sont en général placées sur un plasmide. Un plasmide est une molécule d'ADN circulaire double brin distincte de l'ADN chromosomique, capable de réplication autonome et non essentielle à la survie d'une cellule. Les plasmides sont en général présents en plusieurs copies dans une bactérie. Cela à l'avantage d'amplifier le signal lorsque l'on mesure l'expression d'un gène. Cependant, un désavantage est la possible variation du nombre de copies du plasmide au sein d'une même population ce qui peut introduire un biais dans les études à l'échelle de la cellule. Une solution pour éviter ce problème consiste à intégrer la fusion transcriptionnelle directement dans le chromosome par recombinaison homologue. Notez qu'on perd dans ce cas l'amplification du signal.
Pour construire une fusion transcriptionnelle, on utilise des techniques de biologie moléculaire. On amplifie par PCR le promoteur d'intérêt puis on clone ce promoteur en amont du gène rapporteur sur un plasmide. La première étape consiste à réaliser la construction in silico1. Pour cela j'utilise un logiciel gratuit Gentle mais il existe surement des logiciels plus performants. La vidéo ci-dessous décrit les étapes in silico, réalisées sous Gentle, permettant de placer (cloner) un promoteur en amont d'un gène rapporteur. J'ai réalisé cette démonstration pour montrer à un chercheur peu familier avec la biologie que les séquences d'ADN en " ATCG" ne sont pas uniquement une abstraction: on les manipule vraiment exactement comme on manipulerait un texte en utilisant les fonctions "couper" et "coller". Une fois, la réalisation in silico réussie, on peut se lancer dans la réalisation réelle. Vous vous demandez peut être à quoi ressemble une étape de coupage ou de collage d'ADN? Grossièrement, si vous observiez un biologiste faire de la biologie moléculaire, vous auriez l'impression de voir un homme ou une femme mélanger "de l'eau avec de l'eau" dans un tube pendant plusieurs jours. Puis quelques jours, quelques semaines ou quelques mois plus tard, il sauterait de joie au plafond juste après avoir reçu un résultat de séquençage en criant: ça y est, je l'ai !
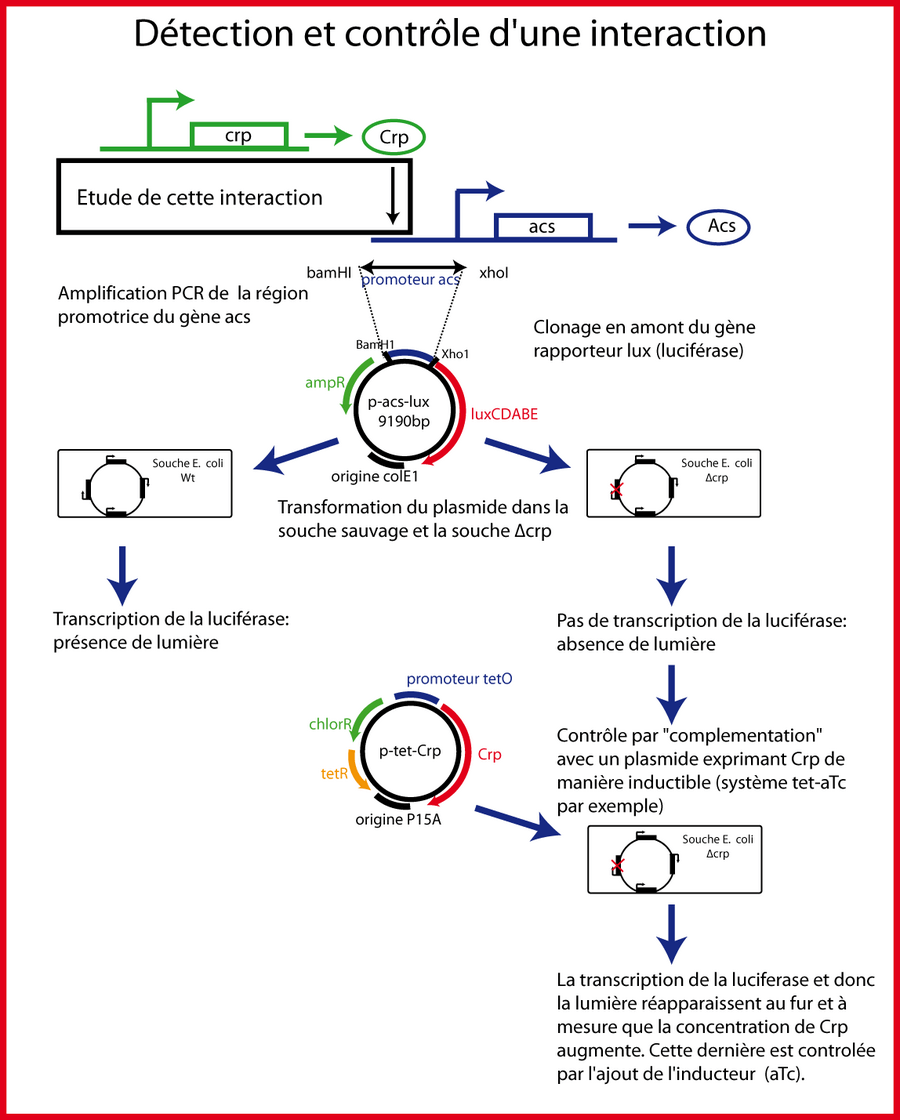
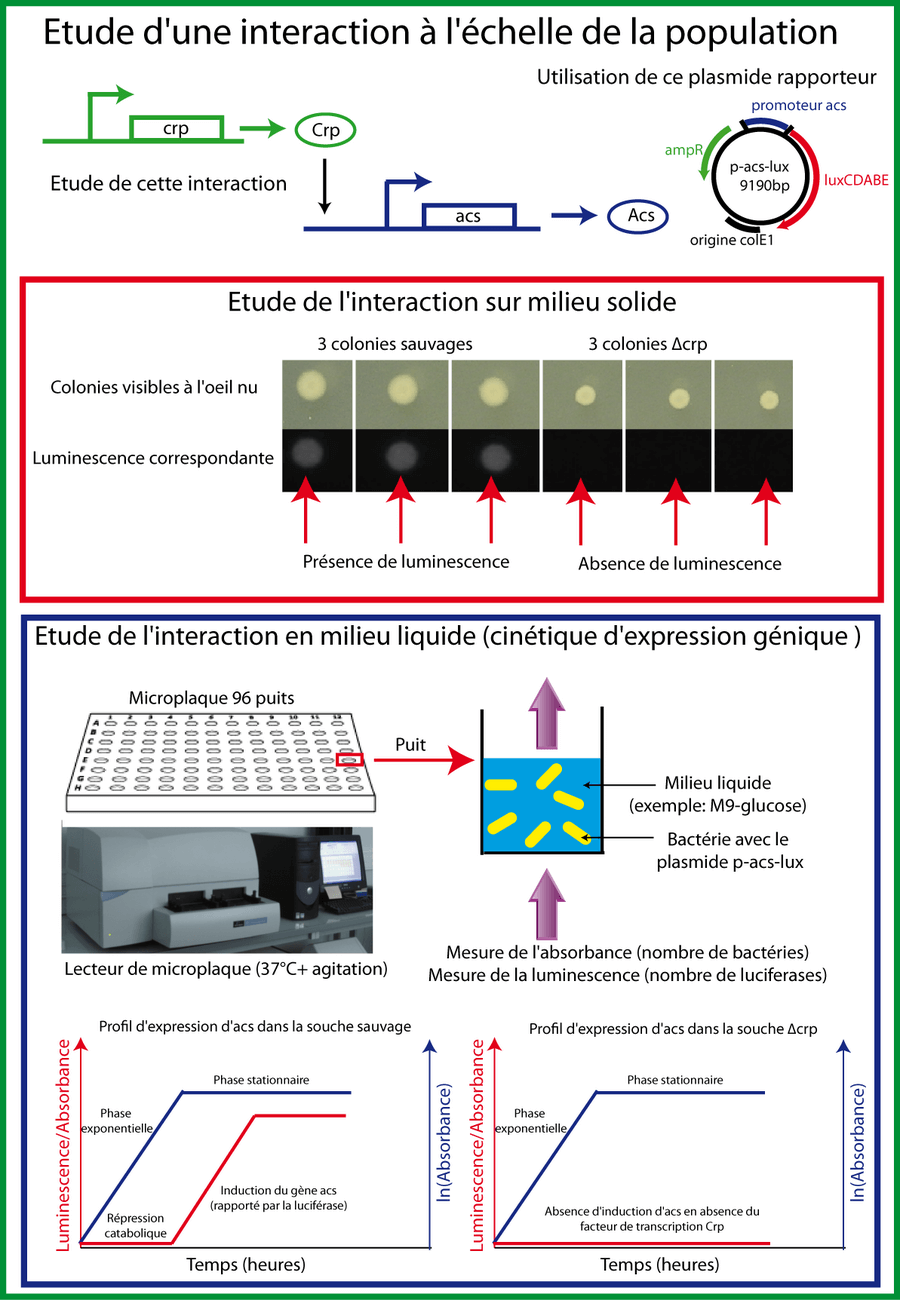
Les fusions transcriptionnelles permettent la mesure de la concentration du rapporteur ou de l'activité du promoteur directement à l'échelle de la colonie, en population (milieu liquide) ou à l'échelle de la cellule (sous le microscope). Lorsque nous mesurons l'activité d'un promoteur dans une population en milieu liquide, nous utilisons un lecteur de microplaque pour mesurer la fluorescence, la luminescence ou l'absorbance. L'intensité I détectée par le photomultiplicateur, après soustraction du bruit de fond, est proportionnelle à la concentration de luciférase ou de GFP dans le puits. L'absorbance mesurée A, après soustraction du bruit de fond, est proportionnelle au nombre de bactéries dans le puits. La concentration moyenne du rapporteur [GFP] ou [Luciferase] dans une bactérie est donc:
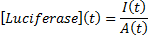
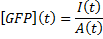
Notez cependant que la luciferase a également besoin d'énergie pour émettre des photons. Quand l'énergie devient limitante, l'émission de photons chute brutalement et ne reflète plus la concentration de l'enzyme luciferase. Cet artefact peut être d'une part contrôlé et d'autre part utilisé pour inférer un certain nombre de paramètre dans la cellule (voir la publication relative à ce phénomène dans cette thèse).
Nous avons écrit un programme pour analyser des données d'expression génique issues de gènes rapporteurs. Ce programme, appelé Wellreader, a fait l'objet d'une publication (Boyer et al.). Il est possible, à partir des données collectées, de remonter à l'activité du promoteur F à l'aide de l'équation ci-dessous (démonstration dans la publication).
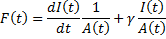
où γ représente le taux de dégradation de la protéine rapporteur.
Pour mesurer une interaction avec une fusion transcriptionnelle, on compare la concentration du rapporteur dans une souche sauvage et dans une souche mutante déletée du facteur de transcription suspecté de contrôler l'activité du promoteur étudié. Notez qu'une différence d'expression ne signifie pas forcement que l'interaction est directe. Enfin pour s'assurer que l'interaction détectée n'est pas artefactuelle, on complémente la souche mutante avec un plasmide exprimant le gène déleté. Si la présence de ce plasmide permet de rétablir le niveau d'expression du mutant au niveau de la souche sauvage alors l'interaction ---directe ou indirecte--- est bien réelle.
Une publication présente dans cette thèse propose une méthode à haut-débit, basée sur les fusions transcriptionnelles, permettant de mesurer l'activité d'un promoteur dans les 4000 mutants d'E. coli.
Les méthodes bioinformatiques
Inférence de la topologie des réseaux de régulation
Toutes les techniques ci-dessus permettent de déterminer expérimentalement une interaction. Cependant, elles sont souvent assez chères et longues à mettre en place. L'apparition de grands "set" de données à haut-débit issues des puces à ADN a permis d'utiliser des méthodes "computationnelles" pour "prédire" des interactions géniques. Une manière simple d'exploiter les données issues de puces à ADN consiste à regrouper les gènes ayant des profils d'expression similaires: c'est la clusterisation. Mais il est également possible de mener l'exploration des données plus en profondeur et d'inférer la topologie du réseau, autrement dit de "reconstruire" ce réseau. Brièvement, des méthodes algorithmiques recherchent des patterns de corrélation ou de probabilités conditionnelles qui indiquent une influence causale. C'est le cas par exemple des algorithmes basés sur un réseau bayesien (Friedman et al. 2000). L'inférence de réseaux biologiques est un domaine très actif et beaucoup trop vaste pour être abordé ici de manière exhaustive. Citons l'"inferelator", un algorithme qui démontre la faisabilité de reconstruction d'un réseau à l'échelle du génome à partir d'un nombre assez limité de puces à ADN (Bonneau et al. 2006). Citons enfin les travaux de l'équipe de Bernhard Ø. Palsson. Cette équipe reconstruit des réseaux transcriptionnels et métaboliques à l'échelle du génome en utilisant de manière conjointe des séquences annotées, des données à haut-débit et des informations bibliographiques (Covert et al. 2004; Feist et al. 2009).
Je vais maintenant insister un peu plus sur une autre méthode d'inférence d'interaction ADN--protéine: la détection de site consensus par bioinformatique. Mon paragraphe reprend en partie un exemple proposé par Hamid Bolouri dans son ouvrage Computational Modelling Of Gene Regulatory Networks: A Primer. Le lecteur pourrait se demander pourquoi je choisis de passer tant de temps à décrire la détermination d'une "séquence--logo". Il y a deux raisons: premièrement, cela me permet de proposer un petit exercice/exemple de programmation sous Matlab. Deuxièmement, cela me permet d'introduire la théorie de l'information de Shannon sur laquelle je compte revenir plus tard. Le lecteur qui craint les détails peut passer au chapitre suivant sans crainte.
Détection de site consensus par bioinformatique
La liaison ADN--protéine est principalement basée sur le pattern des donneurs et des accepteurs de liaison d'hydrogène exposée dans les sillons de la double hélice d'ADN. Ce pattern est donc très dépendant de la séquence et doit être plus ou moins complémentaire d'un pattern similaire présent au niveau du site de liaison de la protéine. La séquence qui maximise l'affinité d'un facteur de transcription donné est appelée séquence consensus. Cette séquence est déterminée expérimentalement grâce à un set de tous les sites séquencés que fixe un facteur de transcription donné. C'est la séquence qui, à chaque position, montre la paire de base la plus souvent trouvée parmi le set. L'utilisation des sites consensus mène les scientifiques dans un piège épistémologique leur faisant confondre le modèle de la réalité (le site consensus) avec la réalité (le site de liaison). En effet, le consensus strict est très éloigné du centre de la distribution gaussienne de l'affinité des sites de liaisons pour un facteur de transcription donné. Le consensus strict est donc très rarement observé. L'affinité trop forte d'un facteur de transcription pour son site consensus pourrait empêcher son détachement et résulterait en une activation ou répression constante du promoteur. Ainsi les algorithmes de recherche de sites de liaison sur une séquence donnée n'utilisent pas un site consensus mais une matrice décrivant pour chaque position, la fréquence d'apparition de chaque nucléotide. Ces matrices assument l'indépendance de chaque base les unes par rapport aux autres (matérialisée dans l'équation par une somme). En d'autre terme, la fréquence avec laquelle une protéine se fixe à un nucléotide donné est proportionnelle à l'affinité de cette protéine pour ce nucléotide.
Supposons qu'un facteur de transcription ait été rapporté pour se lier à un motif de 6 pb grâce à 20 observations expérimentales. Une matrice de positions peut résumer les donnés expérimentales (figure 11; matrice 1).
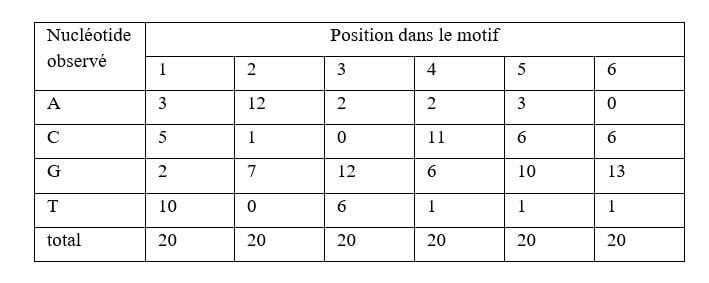
On peut aussi représenter les observations comme des probabilités empiriques en divisant la fréquence de chaque nucléotide par 20 (figure 11; matrice 2). La distribution des fréquences de nucléotide chez un organisme donné est rarement uniforme. On normalise donc la probabilité d'observer un nucléotide dans un site de liaison P(observé) avec la probabilité d'observer un nucléotide dans une séquence "bruit de fond" P(bruit)(figure 11; matrice 3).
Pour calculer la probabilité totale qu'une séquence observée de quelques nucléotides représente un site de liaison, il faut multiplier les probabilités normalisées de chaque paire de base. Sachant que log(A.B)=log(A)+log(B), il est pratique de transformer les probabilités normalisées sous forme logarithmique. Ainsi pour estimer dans quelle mesure une séquence observée représente un site de liaison, on fait simplement la somme log(P(observé)/P(bruit)) à chaque nucléotide. Un programme de prédiction fouillera le génome et sélectionnera, pour une séquence donnée, les signaux de longueur six dont le score est supérieur à un certain seuil (Kel et al. 2003).
Cliquer pour voir le code Matlab
% Ce script est un peu difficile (ne vous cassez pas la tête dessus)
% mais il permet d'identifier comment on stoc des données dans une matrice,
% comment on utilise les fonctions de programmation de base et comment on
% utilise les facilités de plot.Notez qu'il existe une fonction plus
% pratique dans la bioinformatic toolbox pour plotter une sequence-logo
% Fréquence d'apparition des A
%(sur la première ligne), puis C, G et T sur une
% séquence de 6 pb et un set de 20 sequences
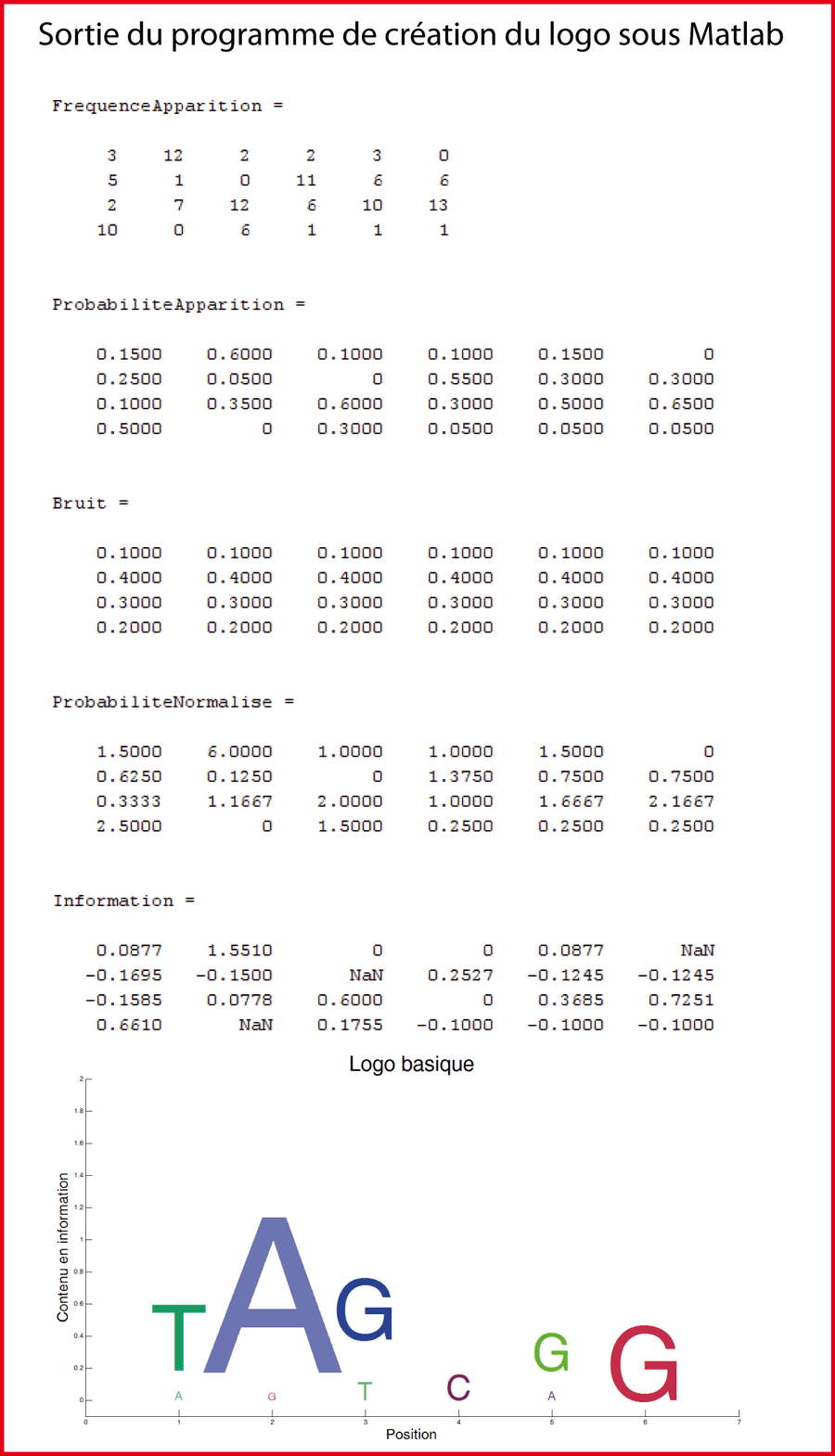
FrequenceApparition= [3 12 2 2 3 0; 5 1 0 11 6 6;...
2 7 12 6 10 13; 10 0 6 1 1 1]
% passage en probabilité en divisant par 20
ProbabiliteApparition=FrequenceApparition/20
% probabilité de chaque paire de base
%à une position donnée (bruit de fond)
Bruit=[0.1 0.1 0.1 0.1 0.1 0.1 ; 0.4 0.4 0.4 0.4 0.4 0.4;...
0.3 0.3 0.3 0.3 0.3 0.3 ;0.2 0.2 0.2 0.2 0.2 0.2]
% normalisation des probabilités
%d'apparition en fonction du bruit
ProbabiliteNormalise=ProbabiliteApparition./Bruit
% calcul de l'information et
%donc de la hauteur des lettres
Information=ProbabiliteApparition .*...
log2(ProbabiliteNormalise)
% on se debarasse des valeurs
%inférieur à 0 et on ordonne les données
[NumBase,Position]=find(Information>0);
HauteurLettre=Information(Information>0);
HauteurLettreOrdonne=[];
BaseOrdonne=[];
for i= 1:6
[Ordonne,Indice]=sort(HauteurLettre(Position==i));
Base=NumBase(Position==i);
HauteurLettreOrdonne=[HauteurLettreOrdonne; Ordonne];
BaseOrdonne=[BaseOrdonne; Base(Indice)];
end
% parametre de visualisation de la figure
f=figure;
set(f,'Color',[1 1 1])
axis([0 7 -0.1 2])
xlabel('Position','FontSize',20)
ylabel('Contenu en information','FontSize',20)
title('Logo basique','FontSize',30)
hold on
% puis on affiche les lettres
%en fonction de leur contenu en information
ParametrePosition=[0 0 0 0];
for i=1:length(HauteurLettreOrdonne)
if Position(i)>1 && Position(i)>Position(i-1)
ParametrePosition(4)=0;
end
switch BaseOrdonne(i)
case 1
ValeurBase='A';
case 2
ValeurBase='C';
case 3
ValeurBase='G';
case 4
ValeurBase='T';
end
Textlogo=text(Position(i),ParametrePosition(4),...
ValeurBase, 'HorizontalAlignment'...
,'center','VerticalAlignment',...
'baseline','FontUnits','centimeters'...
,'FontSize',7*HauteurLettreOrdonne(i),...
'Color',[rand(1) rand(1) rand(1)]);
ParametrePosition=get(Textlogo,'Extent');
end
On peut aussi utiliser les matrices de positions pour créer des "séquences--logo" (figure 11). Ces dernières offrent une manière simple et intuitive pour visualiser le contenu en information de la matrice de positions. Une pile de lettres est utilisée pour indiquer la fréquence d'occurrence de chaque nucléotide à chaque position dans la séquence alignée (axe des abscisses). La lettre la plus fréquente est placée en première position et les autres sont empilées dessous dans l'ordre des fréquences décroissantes.
La théorie de l'information nous fournit une mesure du contenu en information (IC) à chaque position i donné par la formule suivante (Workman et al. 2005) où b représente les 4 bases A, T, C ou G.
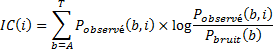
Le calcul de la hauteur d'une lettre est simplement
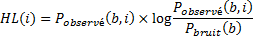
La hauteur totale à une position donnée indique le contenu en information totale de cette position. Pour un site de liaison candidat donné avec un bruit de fond uniformément distribué de nucléotides aléatoires (0.25), une hauteur de 2 à n'importe quelle position dans la séquence du logo signifie qu'il n'y a qu'un seul nucléotide qui peut apparaitre à cette position.
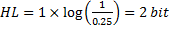
A l'inverse, une hauteur de 0 signifie que chaque nucléotide a une probabilité équivalente de se retrouver à cette position.
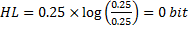
L'utilisation du logarithme base 2 permet de quantifier l'information en bit, c'est-à-dire un nombre binaire qui n'a pour valeur que 0 ou 1. En utilisant 2 bits, on peut indiquer à une position donnée si le nucléotide est un A, un T, un C ou un G. C'est une autre manière de dire que 2 bits permettent 22=4 combinaisons de valeurs.
Souvent les séquences logo font apparaitre deux zones à fort contenu en information entrecoupés d'une zone à plus faible contenu en information. Les deux zones à fort contenu en information correspondent au sillon majeur de l'ADN où le facteur de transcription peut aisément discriminer les bases ce qui est moins le cas dans le sillon mineur. Ceci est un bel exemple pour entrevoir la structuration physique en double hélice de l'ADN.
Bibliographie
- Bonneau, R., D. J. Reiss, et al. (2006). "The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo." Genome Biol 7(5): R36.
- Boyer, F., B. Besson, et al. "WellReader: a MATLAB program for the analysis of fluorescence and luminescence reporter gene data." Bioinformatics 26(9): 1262-3.
- Bray, D., M. D. Levin, et al. (2007). "The chemotactic behavior of computer-based surrogate bacteria." Curr Biol 17(1): 12-9.
- Covert, M. W., E. M. Knight, et al. (2004). "Integrating high-throughput and computational data elucidates bacterial networks." Nature 429(6987): 92-6.
- Feist, A. M., M. J. Herrgard, et al. (2009). "Reconstruction of biochemical networks in microorganisms." Nat Rev Microbiol 7(2): 129-43.
- Friedman, N., M. Linial, et al. (2000). "Using Bayesian networks to analyze expression data." J Comput Biol 7(3-4): 601-20.
- Gama-Castro, S., V. Jimenez-Jacinto, et al. (2008). "RegulonDB (version 6.0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation." Nucleic Acids Res 36(Database issue): D120-4.
- Grainger, D. C. and S. J. Busby (2008). "Methods for studying global patterns of DNA binding by bacterial transcription factors and RNA polymerase." Biochem Soc Trans 36(Pt 4): 754-7.
- Kel, A. E., E. Gossling, et al. (2003). "MATCH: A tool for searching transcription factor binding sites in DNA sequences." Nucleic Acids Res 31(13): 3576-9.
- Keseler, I. M., C. Bonavides-Martinez, et al. (2009). "EcoCyc: a comprehensive view of Escherichia coli biology." Nucleic Acids Res 37(Database issue): D464-70.
- Kolb, A., S. Busby, et al. (1993). "Transcriptional regulation by cAMP and its receptor protein." Annu Rev Biochem 62: 749-95.
- Kremling, A., S. Kremling, et al. (2009). "Catabolite repression in Escherichia coli- a comparison of modelling approaches." Febs J 276(2): 594-602.
- Miller, J. H. (1972). "Experiments in Molecular Genetics." Cold Spring Harbor
- Monod, J. (1942). "Recherches sur la croissance des cultures bactériennes." 2nd edn. Paris:Hermann
- Postma, P. W., J. W. Lengeler, et al. (1993). "Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria." Microbiol Rev 57(3): 543-94.
- Saier, M. H., Jr. and M. Crasnier (1996). "Inducer exclusion and the regulation of sugar transport." Res Microbiol 147(6-7): 482-9.
- Shimomura, O., F. H. Johnson, et al. (1962). "Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea." J Cell Comp Physiol 59: 223-39.
- Vladimirov, N. and V. Sourjik (2009). "Chemotaxis: how bacteria use memory." Biol Chem 390(11): 1097-104.
- Wang, Z., M. Gerstein, et al. (2009). "RNA-Seq: a revolutionary tool for transcriptomics." Nat Rev Genet 10(1): 57-63.
- Workman, C. T., Y. Yin, et al. (2005). "enoLOGOS: a versatile web tool for energy normalized sequence logos." Nucleic Acids Res 33(Web Server issue): W389-92.
Notes de bas de page
- in silico: Réalisé dans le "silicium" d'un ordinateur par opposition à in vitro et in vivo. Ce terme a été introduit par Antoine Danchin dans les années 1990.