Réseaux de régulation chez Escherichia coli
Résumé
L'adaptation d'une bactérie aux changements de son environnement est contrôlée par un réseau de régulation large et complexe, faisant intervenir de nombreux acteurs et modules différents. Dans ce travail, nous avons étudiés un module de régulation spécifique, contrôlant l'adaptation de la bactérie Escherichia coli à un changement de sources de carbone. Dans un milieu contenant du glucose et de l'acétate, la croissance est divisée en deux phases : les bactéries utilisent préférentiellement le glucose et commencent à métaboliser l'acétate qu'après l'épuisement du glucose. En effet, la présence du glucose réprime la transcription d'un gène nécessaire à la croissance sur acétate, le gène acs (codant pour l'acétyl-CoA synthétase). Le mécanisme régulateur fait intervenir le facteur de transcription Crp-AMPc et le système de transfert de phosphate (PTS), qui permet l'import du glucose.
Plusieurs modèles décrivent en détail la cascade de réactions moléculaires à l'origine de cette "répression catabolique". Cependant, certaines de nos observations expérimentales ne sont pas correctement prédites par les modèles actuels. Ces modèles doivent être révisés ou complétés. L'outil majeur que nous employons pour les expériences est la fusion transcriptionnelle : une région promotrice fusionnée en amont d'un gène rapporteur (GFP, luciferase). Avec ces constructions, nous mesurons la dynamique de l'expression génique dans différentes souches (mutants) et différentes conditions environnementales. Les observations à l'échelle de la population sont corroborées par des mesures similaires à l'échelle de la cellule unique. Nous utilisons cette même technologie pour construire de petits systèmes synthétiques qui sondent davantage le phénomène de répression catabolique. Nous avons ainsi créé un interrupteur génétique dont le fonctionnement est contrôlé par le flux glycolytique et nous avons construit un petit système de communication intercellulaire basé sur la molécule AMPc. Enfin, nous proposons une manière originale de mesurer l'état métabolique des cellules en utilisant la dépendance énergétique de la luciferase.
Introduction
La biologie des systèmes
La biologie des systèmes cherche à intégrer les différents niveaux d'information qui structurent un organisme. Ces niveaux d'information s'établissent à différentes échelles : macroscopique comme les mécanismes physiologiques, microscopique comme la communication cellulaire ou nanométrique comme la régulation génique. Interconnecter ces différents niveaux permettra, à terme, de comprendre, de prédire, voire de contrôler la dynamique de fonctionnement global d'un système vivant. La difficulté d'une telle approche vient du fait que chaque niveau est régi par des lois qui lui sont propre. Ainsi, avant de pouvoir construire un modèle global multi-échelle d'un système vivant complexe, il est nécessaire de diviser les difficultés en se focalisant à un niveau d'information donné sur un organisme suffisamment simple. Dans cette étude, nous nous focaliserons sur la dynamique de fonctionnement du réseau de régulation d'un organisme très étudié et facilement manipulable : la bactérie Escherichia coli.
L'adaptation d'une bactérie aux changements dans son environnement est contrôlée au niveau moléculaire par un réseau de régulation large et complexe impliquant des gènes, des ARN, des protéines et des métabolites. Transduction du signal, régulations transcriptionnelles, flux métaboliques sont étroitement interconnectés même si ils fonctionnent à des échelles de temps très différentes (de la milliseconde au jour). Connaître la topologie de ce réseau de régulation est le premier objectif de la biologie des systèmes. La base de données Ecocyc liste plus de 5345 interactions régulatrices chez E. coli et ce nombre ne cesse d'augmenter (Keseler et al., 2010). Etudier le fonctionnement dynamique de ce réseau est le second objectif de la biologie des systèmes. Cette dynamique est non linéaire et comprend de nombreuses boucles de rétroaction. De plus, la nature intrinsèquement stochastique des mécanismes de régulation (mouvement brownien) rend le réseau de régulation idiosyncratique : pour un même génotype, une topologie du réseau identique et une même perturbation, deux bactéries peuvent adopter deux phénotypes distincts lors de l'adaptation (Veening et al., 2008).
En l'état actuel des connaissances, il n'est pas encore possible de comprendre et de contrôler finement la dynamique de fonctionnement de cet énorme réseau de régulation. Nous nous sommes donc focalisés sur un module de régulation plus restreint mais faisant intervenir à la fois une réorganisation de la transcription et une réorganisation des flux métaboliques : nous avons choisi, comme point de départ, d'étudier l'adaptation de la bactérie E. coli à un changement de sources de carbone : du glucose à l'acétate (Wolfe, 2005).
Le système d'étude
Lorsque les bactéries E. coli sont placées dans un milieu contenant du glucose, elles consomment ce glucose tout en secrétant de l'acétate dans le milieu extérieur. Une fois le glucose épuisé, les bactéries réimportent cet acétate et l'utilisent comme source d'énergie. Cette transition glucose-acétate entraîne une inversion des flux métaboliques (de la glycolyse à la néoglucogenèse) et une profonde réorganisation de l'expression des gènes (Oh et al., 2002).
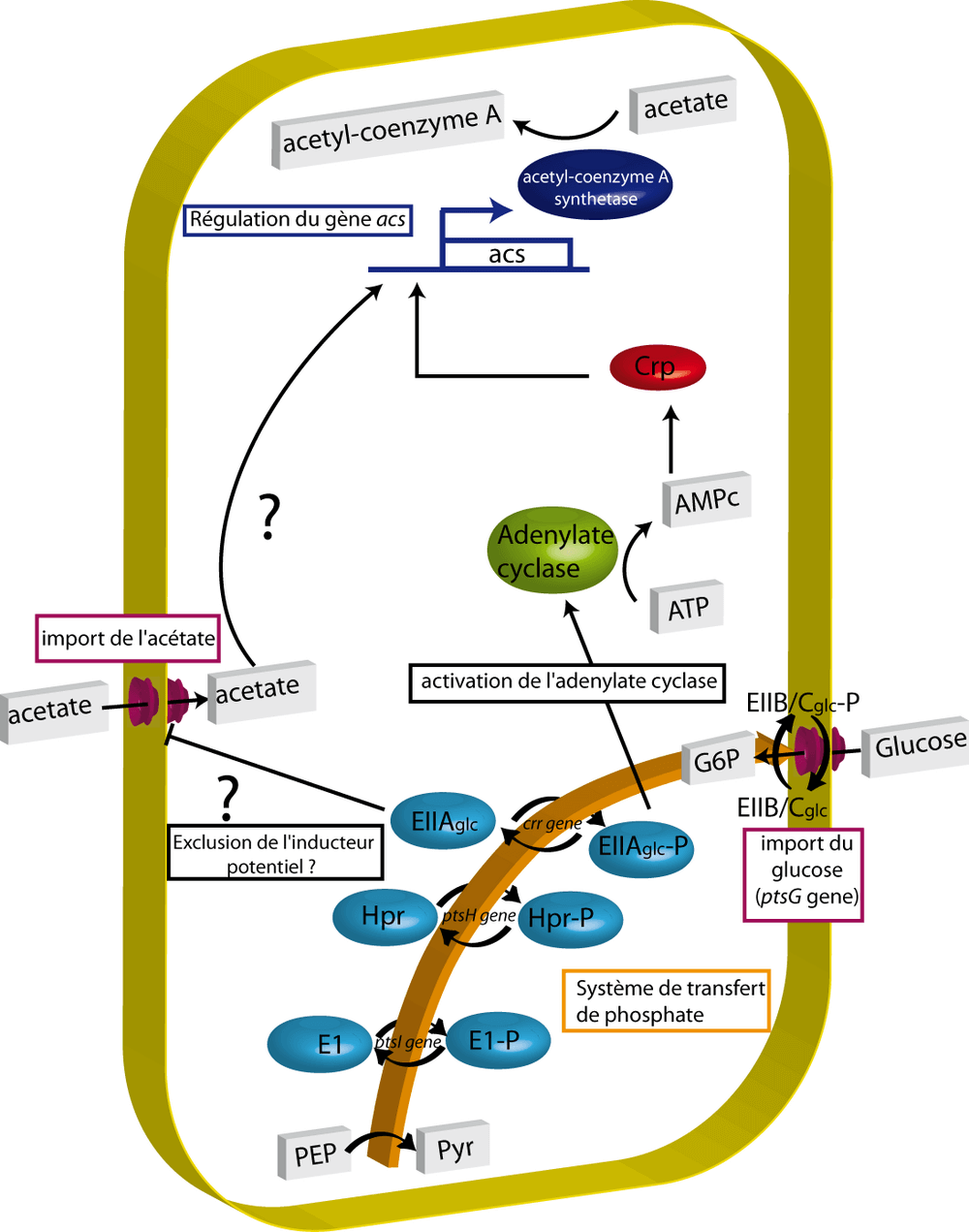
Pour pouvoir utiliser l'acétate comme source d'énergie principale, il est nécessaire de le convertir en acétyl-coenzyme A. Cette première étape est réalisée par l'acétyl-coenzyme A synthétase encodée par le gène acs (Kumari et al., 1995). Ce gène a un rôle précoce et prédominant dans l'établissement de la transition glucose-acétate c'est pourquoi nous nous sommes focalisés sur la régulation de son expression. En effet, sa transcription est connue pour être activée par le facteur de transcription Crp qui n'est actif que lorsque le glucose est épuisé (Kumari et al., 2000). Ce mécanisme, appelé répression catabolique, fait intervenir une petite molécule : l'adénosine monophosphate cyclique (AMPc). Selon un modèle classique, lorsque le glucose est épuisé, une cascade de réactions aboutit à l'activation d'une enzyme, l'adenylate cyclase, qui produit alors une forte concentration d'AMPc. Cette molécule se fixe à la protéine Crp et change sa conformation. Le complexe ainsi formé a une plus grande affinité pour son site de liaison à l'ADN, au niveau de la séquence promotrice du gène acs. La fixation de ce complexe Crp-AMPc entraine l'activation de la transcription de ce gène.
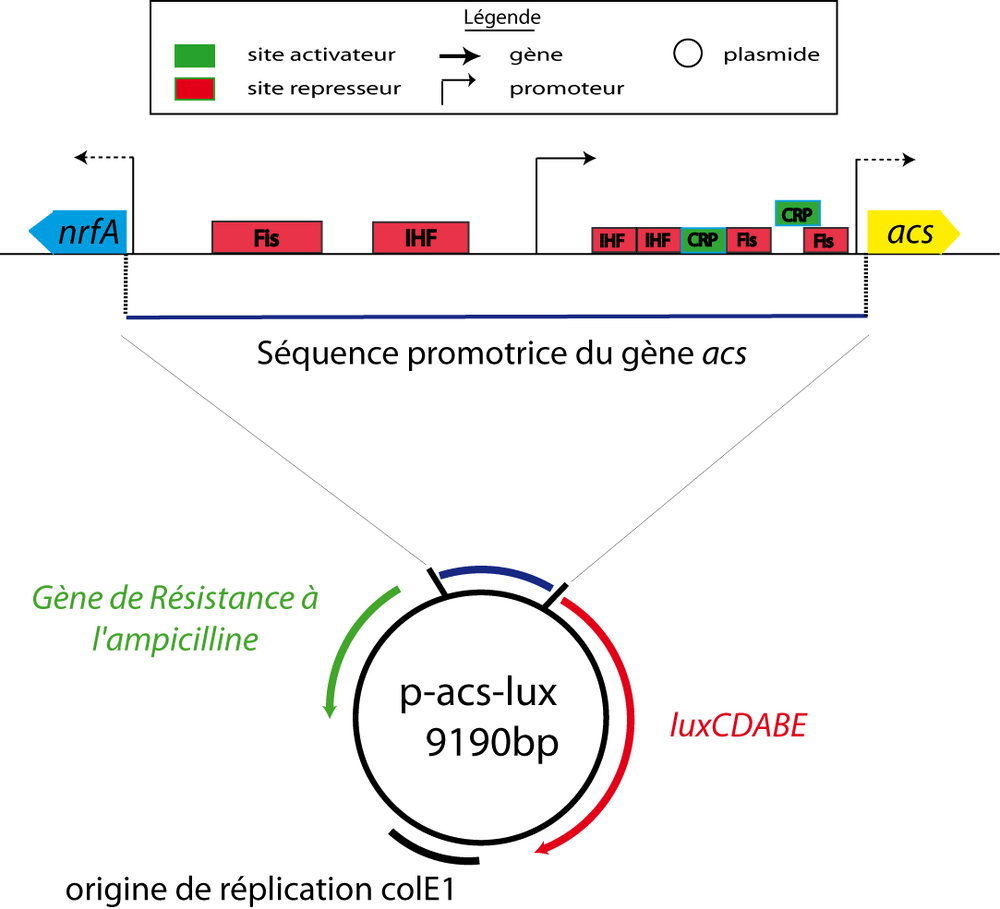
Pour pouvoir mesurer l'expression du gène acs avec une haute résolution temporelle, nous utilisons la technologie des fusions transcriptionnelles. Nous fusionnons, sur un plasmide multicopie, la séquence promotrice du gène acs en amont d'un gène rapporteur (l'operon luxCDABE) codant pour la luciferase : cet outil serait désormais appelé p-acs-lux (Figure 1). Ainsi l'activité du promoteur acs est directement reliée à la concentration de luciferase qui est, elle-même, proportionnelle à la production de lumière, facilement mesurable
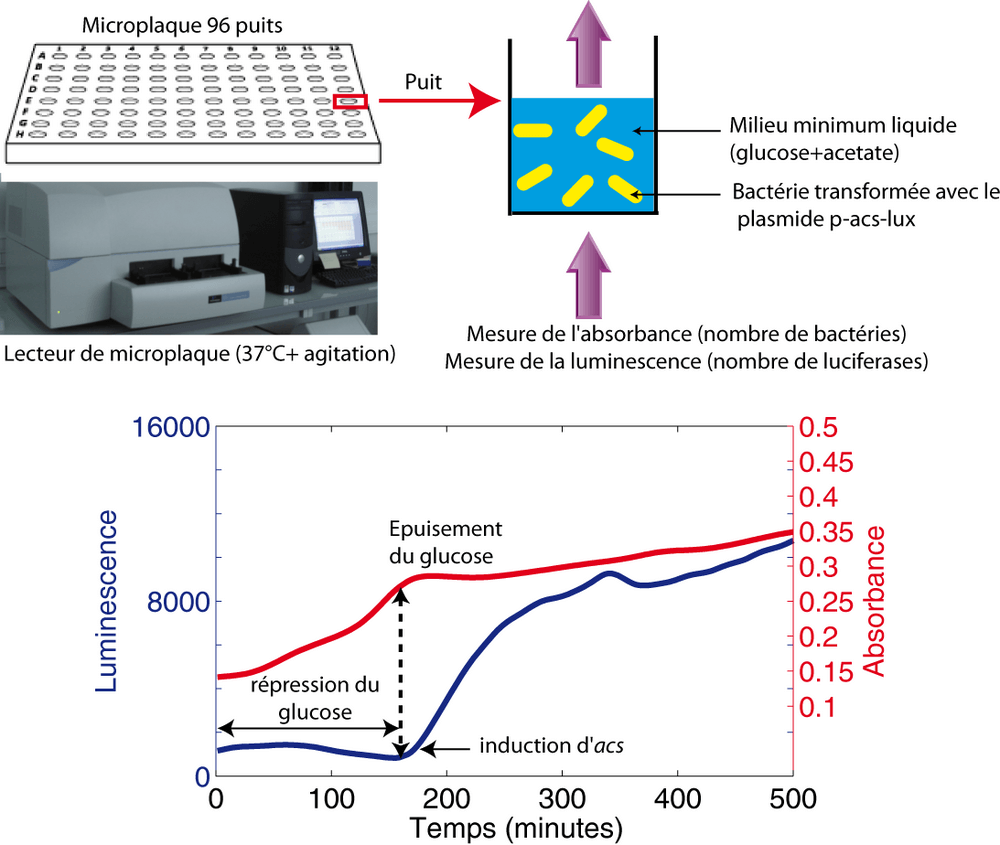
Une fois notre outil construit, nous l'avons placé, par transformation, dans des bactéries E. coli sauvages et nous avons fait pousser ces dernières dans un milieu minimum contenant du glucose et de l'acétate. La croissance a lieu dans un lecteur de microplaque ce qui nous permet de mesurer, à intervalle de temps régulier, l'absorbance qui reflète le nombre de bactéries et la luminescence qui reflète l'activité du promoteur acs. Avec cette méthode, nous avons pu observer que l'activité du promoteur acs est fortement réprimée durant la croissance sur glucose, puis augmente brutalement lorsque ce dernier est épuisé (Figure 2). Pour s'assurer que ce profil d'expression est correct, nous l'avons contrôlé à l'aide d'une autre fusion transcriptionnelle composée d'un autre rapporteur : la gfp. Le profil obtenu en fluorescence est similaire à celui obtenu en luminescence. Enfin, pour définitivement exclure tout artefact, nous avons également confirmé ce profil à l'aide d'une méthode totalement indépendante : la qRT-PCR.
Logique de régulation de l'expression d'acs
Dans les paragraphes qui suivent, nous allons nous intéresser à la cascade d'événements qui a lieu entre l'épuisement du glucose et l'activation du promoteur acs. C'est en effet cette cascade d'événements qui est en mesure d'expliquer l'allure du profil d'expression décrit précédemment.
La littérature décrit l'activation de l'expression du gène acs par le facteur de transcription Crp. Nous avons vérifié cette information en mesurant son expression dans un mutant Δcrp. Comme prévu, dans une telle souche, il n'y a aucune expression du gène acs. Ceci entraîne un phénotype facile à observer : l'absence de croissance sur acétate. En réintroduisant, dans ce mutant, une copie du gène crp, on retrouve l'expression du gène acs et la croissance sur acétate. Les choses se passent de manière très similaire dans un mutant ΔcyaA, c'est à dire dans un mutant sans adenylate cyclase. En absence d'AMPc, le facteur de transcription Crp ne peut pas se lier à la séquence promotrice du gène acs et ce dernier n'est pas exprimé. A l'inverse, l'ajout d'AMPc exogène dans le milieu rétablit l'expression du gène acs.
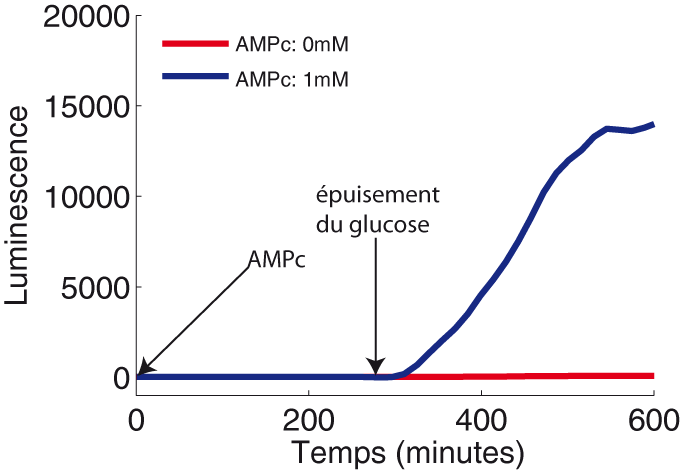
Le résultat de cette dernière expérience a mis en évidence une divergence vis-à-vis du modèle classique de la répression catabolique. L'ajout d'AMPc dans le milieu aurait dû contourner la répression du glucose et induire directement l'expression du gène acs malgré la présence de glucose. Or ce n'est pas ce que nous observons: l'expression d'acs dans le mutant ΔcyaA est complémentée par l'ajout d'AMPc exogène, mais seulement après épuisement du glucose (Figure 3). Nous avons donc déduit qu'il existait un autre mécanisme régulateur empêchant l'expression d'acs tant qu'il y a du glucose dans le milieu.
Un criblage à haut-débit pour détecter de nouvelles interactions géniques
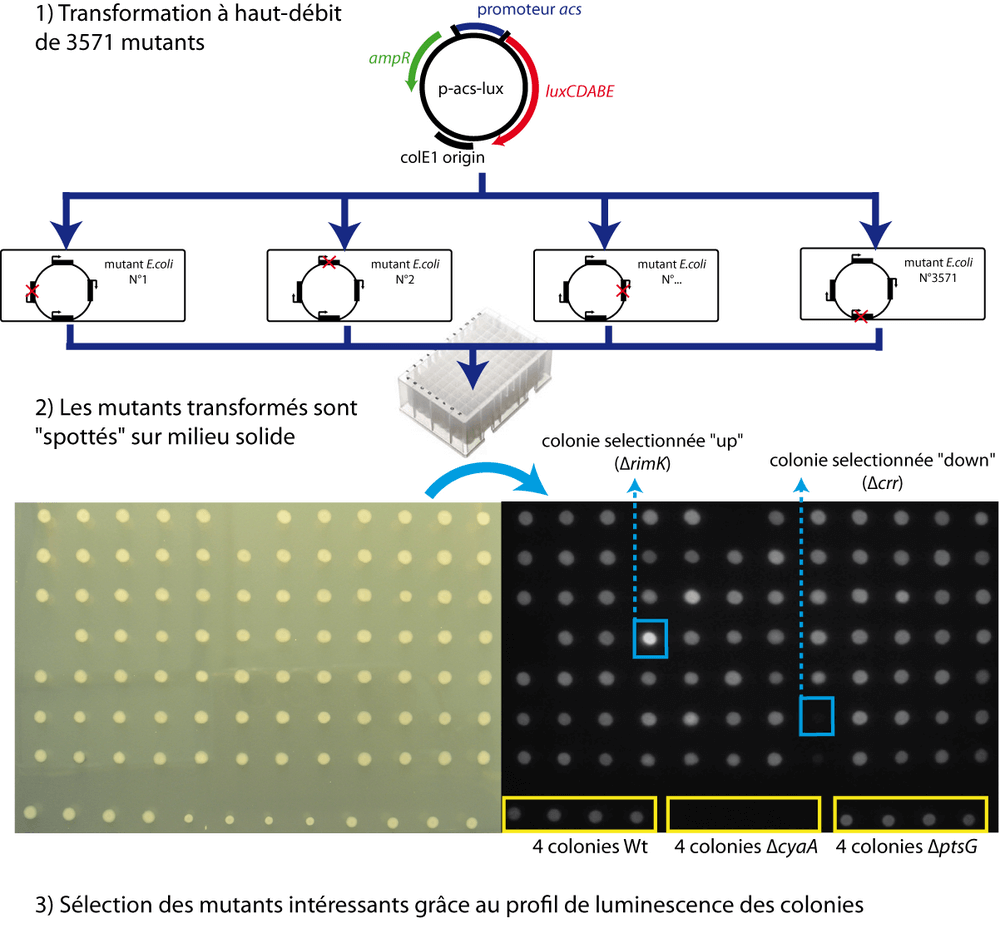
Pour rechercher ce mécanisme de régulation inconnu, nous avons développé un nouveau type de criblage à haut-débit. Nous avons transformé les 4000 simples mutants d'Escherichia coli, provenant d'une collection (Baba et al. 2006), avec notre fusion transcriptionnelle p-acs-lux. Le criblage consiste à comparer la luminescence des colonies mutantes avec celle de la colonie sauvage : une différence significative pouvant suggérer que le gène déleté contrôle, directement ou indirectement, l'expression du gène acs (Figure 4).
Parmi les 4000 mutants, nous avons identifié deux groupes de gènes bien distincts. Le premier groupe est composé de gènes impliqués dans la fabrication de l'énergie. La luciferase, pour émettre des photons, a besoin de pouvoir réducteur ce qui implique la présence d'un métabolisme central fonctionnel (Meighen, 1991). Certaines colonies mutantes, émettant significativement moins de lumière, révèlent un épuisement énergétique plutôt qu'une régulation spécifique sur l'activité du promoteur acs : il s'agit donc de faux positifs. Nous reviendrons plus tard en détail sur cet artefact énergétique pour montrer qu'il peut également être utile.
Un deuxième groupe de gènes a été identifié par notre criblage : les gènes du système de transfert de phosphate (PTS). Les enzymes correspondantes sont impliquées dans l'import et la phosphorylation du glucose pour donner du glucose-6-phosphate. Le phosphate ajouté a subi une cascade de transfert partant de phospho-enol-pyruvate (PEP) et se déplaçant successivement sur l'enzyme E1 (ptsI), puis HPR (ptsH) puis EIIAglc (crr), puis sur le transporteur EIIB/Cglc (ptsG) pour se retrouver enfin sur le 6 ème carbone du glucose formant le glucose-6-phosphate (Postma et al. 1993).
La détection par notre criblage des gènes appartenant au PTS n'est pas étonnante en soi. En effet, l'implication de ce système dans la répression catabolique est connue depuis longtemps. Ce système fait le lien entre l'épuisement du glucose et l'augmentation de la concentration d'AMPc dans la cellule. Le modèle actuel postule que seule la forme phosphorylée de l'enzyme EIIAglc soit capable d'activer l'adenylate cyclase et donc d'augmenter la synthèse d'AMPc. Or la forme phosphorylée d'EIIAglc est la version prédominante en absence de glucose (schéma récapitulatif de la figure 5). Notre criblage n'a pas seulement réussi à faire ressortir les mutants du PTS, il a également révélé de nouvelles subtilités dans le fonctionnement de la répression catabolique. En effet, les colonies Δcrr (sans EIIAglc) et ΔptsI (sans E1) ont été sélectionnées pour leur faible luminescence alors que la colonie ΔptsG (sans EIIB/Cglc) a été sélectionnée sur la base de sa forte luminescence. Ce résultat n'est ni prédictible ni explicable par le modèle ci-dessus.
Un des avantages de notre méthode à haut-débit est d'offrir la possibilité de mener des investigations plus poussées en testant les phénotypes intéressants dans différentes conditions et en dynamique. Par exemple, nous avons pu vérifier que, dans le mutant Δcrr (sans EIIAglc), l'ajout d'AMPc exogène dans le milieu rétablit l'expression du gène acs. Ce résultat suggère (sans le démontrer) le mécanisme d'activation de l'adenylate cyclase par la version phosphorylée de EIIAglc. Pourtant (et comme observé avec le mutant ΔcyaA) dans le mutant Δcrr, l'AMPc rétablit l'activité du promoteur acs seulement une fois que le glucose est épuisé : il subsiste donc un mécanisme de répression indépendant de l'enzyme EIIAglc. Nous allons voir que ce résultat d'expérience contredit un deuxième modèle de la répression catabolique.
Recherche du mécanisme répresseur
Il existe une deuxième explication au phénomène de répression catabolique : le mécanisme d'exclusion de l'inducteur (Saier & Roseman, 1972). Beaucoup de gènes chez E. coli ont leur expression contrôlée par un couple facteur de transcription-inducteur. Citons le couple classique LacI-lactose. Dans le modèle de l'exclusion de l'inducteur, c'est, cette fois, la forme déphosphorylée de EIIAglc qui inhibe l'import du lactose au niveau du transporteur. Or la forme déphosphorylée d'EIIAglc est prédominante en présence de glucose (schéma récapitulatif de la figure 5). L'inhibition de l'import du lactose empêche ce dernier de jouer son rôle d'inducteur.
Dans le cas d'acs, il n'existe pas de facteur de transcription connu qui, en présence d'acétate, activerait ou de-réprimerait l'expression du gène acs. Notre criblage n'a d'ailleurs pas détecté l'existence d'un tel facteur. Il est difficile d'exclure expérimentalement l'hypothèse du rôle "inducteur" de l'acétate car ce dernier est produit et sécrété par la bactérie elle-même. Cependant, si la version déphosphorylée de EIIAglc inhibait l'entrée de l'inducteur potentiel "acétate", alors l'ajout d'AMPc exogène dans le milieu de croissance d'un mutant Δcrr (sans EIIAglc) devrait contourner la répression exercée par le glucose via EIIAglc. Or ce n'est pas ce que nous observons. Ainsi ni le modèle d'exclusion de l'inducteur ni le modèle passant par l'activation de l'adenylate cyclase ne sont en mesure d'expliquer nos données.
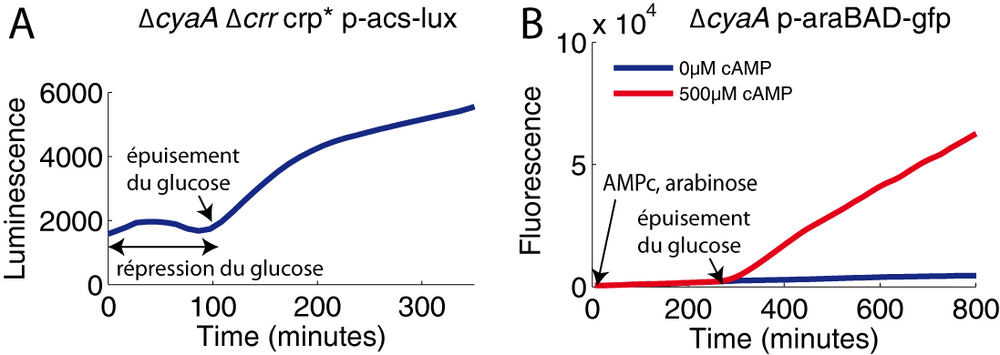
A partir de là, il restait une dernière hypothèse à exclure : l'AMPc exogène que nous ajoutons pourrait ne pas réussir à rentrer dans la bactérie en présence de glucose ce qui pourrait expliquer l'absence de levée de répression. D'autant que la bactérie E. coli secrète des quantités non négligeables d'AMPc dans le milieu extérieur sans que l'on connaisse la fonction de cet AMPc secrété. Ce pool extérieur d'AMPc pourrait être la résultante d'un mécanisme d'import/export servant à contrôler finement la concentration d'AMPc intracellulaire. Pour exclure cette hypothèse, nous avons utilisé une protéine dite Crp*, qui est une copie de Crp légèrement modifiée (Khankal et al., 2009). Cette protéine a la particularité d'être "AMPc indépendante" : elle peut activer les promoteurs Crp-AMPc dépendants sans nécessité la formation du complexe Crp-AMPc. Nous avons donc construit une souche Δcrr ΔcyaA crp*. Quand cette souche pousse dans un milieu avec du glucose et de l'acétate (et sans AMPc exogène), le profil d'expression du gène acs est très similaire à celui de la souche sauvage : la répression du glucose est toujours visible malgré le fait que les deux pierres angulaires des modèles de la répression catabolique, EIIAglc et l'adenylate cyclase, ont été retirées (Figure 6A). L'hypothèse du contrôle de l'import/export de l'AMPc, comme explication de nos résultats, devient caduque car la répression du glucose perdure dans cette souche en absence d'AMPc.
Un phénomène général
Ces résultats obtenus sont-ils spécifiques à la régulation du gène acs ou sont-ils généralisables à l'ensemble des promoteurs Crp-AMPc dépendants ? Pour répondre à cette question, nous avons sélectionné plusieurs fusions transcriptionnelles Crp-AMPc dependantes dans une collection (Zaslaver et al., 2006). Il s'agit des promoteurs araBAD, glpA et sdhC. Bien qu'il existe des subtilités quantitatives, nous avons pu montrer que les résultats obtenues avec acs sont généralisables à ces 3 promoteurs (Figure 6B). Nous pensons donc que nos recherches portent sur le mécanisme général de la répression catabolique.
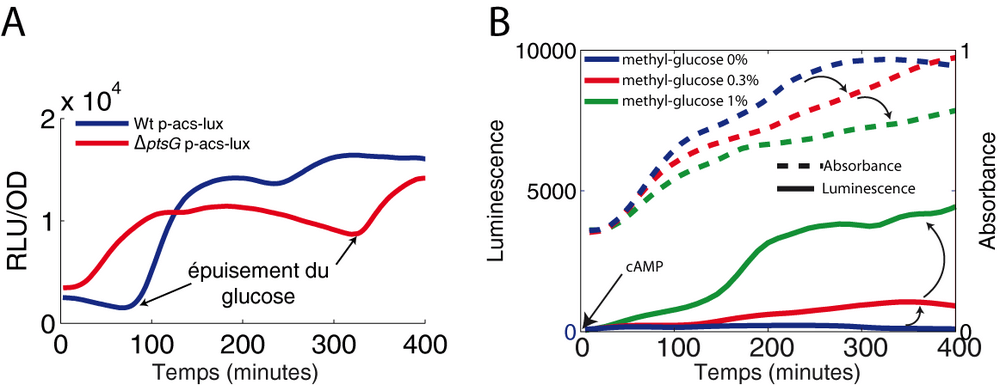
Le flux glycolytique
Dans toutes nos expériences, le paramètre qui semble crucial pour contrôler l'état de la répression catabolique (et donc l'état d'activation du régulon Crp) est l'état du flux glycolytique. En effet, dans le mutant ΔptsG (sans le transporteur du glucose), le niveau basal d'expression d'acs est surélevé par rapport à la souche sauvage car le flux glycolytique y est, à l'inverse, réduit (Figure 7A). Une autre manière pour diminuer le flux glycolytique consiste à utiliser un sucre non métabolisable (l'α-methyl-glucose) agissant en compétition avec le glucose. La littérature décrit ces sucres comme des répresseurs cataboliques alors que nous ne pensons pas qu'ils le soient. Au contraire, lorsque le ratio glucose/α-methyl-glucose génère la réduction adéquate du flux glycolytique, nous avons pu activer l'expression de plusieurs promoteurs Crp-AMPc dépendants (Figure 7B).
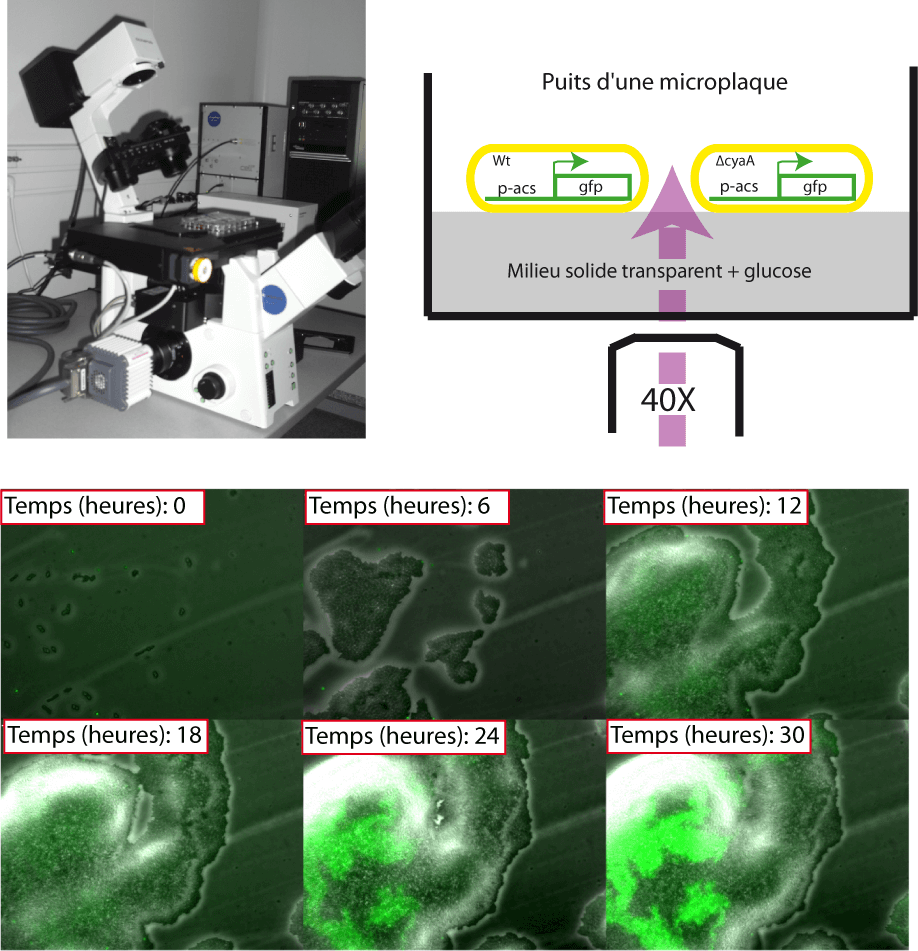
Expériences à l'échelle de la cellule individuelle
Les données présentées jusqu'à maintenant sont principalement issues d'expériences effectuées à l'échelle de la population dans les puits d'une microplaque. Nous avons également vérifié certains de nos résultats à l'échelle de la cellule isolée. Par exemple, nous avons pu mettre en évidence l'induction du gène acs lorsque le glucose est épuisé (Figure 8). De même, nous avons pu observer, sous le microscope, la répression catabolique exercée sur l'expression de l'operon araBAD malgré la présence de l'inducteur (arabinose) et de l'AMPc. L'expression de l'operon araBAD est connue pour amplifier les effets du bruit moléculaire à cause de la présence d'une boucle de rétroaction positive (Megerle et al., 2008). Nous avons également observé ces variations stochastiques. De plus, nous pensons avoir été en mesure de contrôler finement la distribution de l'expression de cet opéron dans la population de cellules en faisant varier le flux glycolytique à l'aide de l'α-methyl-glucose.
Notre système expérimental sous le microscope (milieu solide, formation de microcolonies) est très différent de celui en milieu liquide. Pourtant, les résultats obtenues dans ces deux types de conditions sont similaires ce qui démontrent leur certaine robustesse. Cela peut sembler anodin mais le mécanisme responsable de la répression catabolique fait encore l'objet de nombreux débats (Inada et al., 1996; Crasnier-Mednansky, 2008; Gorke & Stulke, 2008; Narang, 2009). Les désaccords sont souvent liés à l'obtention de résultats différents entre les équipes pour une même expérience : par exemple, l'effet de l'ajout d'AMPc exogène sur le regulon Crp en présence de glucose. Tout le monde s'accorde cependant pour dire que les modèles actuels sont incomplets et doivent être révisés. Sans certitude cependant, nous pensons que le mécanisme responsable de la répression catabolique est encore inconnu. Il ne s'agit, pour nous, ni des variations de concentration de l'AMPc intracellulaire, ni des variations de l'état de phosphorylation d' EIIAglc. Paradoxalement, et c'est ce qui rend les choses si complexes, les rôles du PTS et de l'AMPc sont incontestables.
Utiliser la répression catabolique en biologie synthétique
Ainsi nous pensons que le fonctionnement de l'interrupteur, contrôlé par le flux glycolytique et contrôlant l'activité des promoteurs Crp-AMPc dépendants, reste inconnu. Nous avons créé un petit système synthétique qui illustre la fiabilité de cet interrupteur (Figure 9). Notre système est constitué de deux fusions transcriptionnelles distinctes. La première rapporte l'activité du promoteur uhpT via le rapporteur GFP. Ce promoteur est activé par l'inducteur glucose-6-phosphate (G6P). La deuxième fusion rapporte l'activité du promoteur acs via le rapporteur luciferase. Le mécanisme d'interrupteur fonctionne de la manière suivante : les bactéries poussent sur un milieu minimum contenant du G6P et de l'acétate. La présence du G6P dans le milieu active le promoteur uhpT (ON), ce qui permet l'utilisation de ce sucre comme source de carbone. La mise en place du flux glycolytique empêche l'activité du promoteur acs (OFF). Lorsque le G6P est épuisé, le promoteur uhpT cesse d'être actif (OFF), le flux glycolytique s'effondre ce qui active le promoteur acs (ON). Ce petit système synthétique, bien que basé sur le mécanisme inconnu de la répression catabolique, est robuste, performant et rapide. Nous avons également implémenté une version un peu différente à l'échelle de la cellule isolée. Ce petit détour par la biologie synthétique nous offre une excellente transition pour amorcer la prochaine partie : la description d'un système synthétique de communication intercellulaire par l'AMPc.
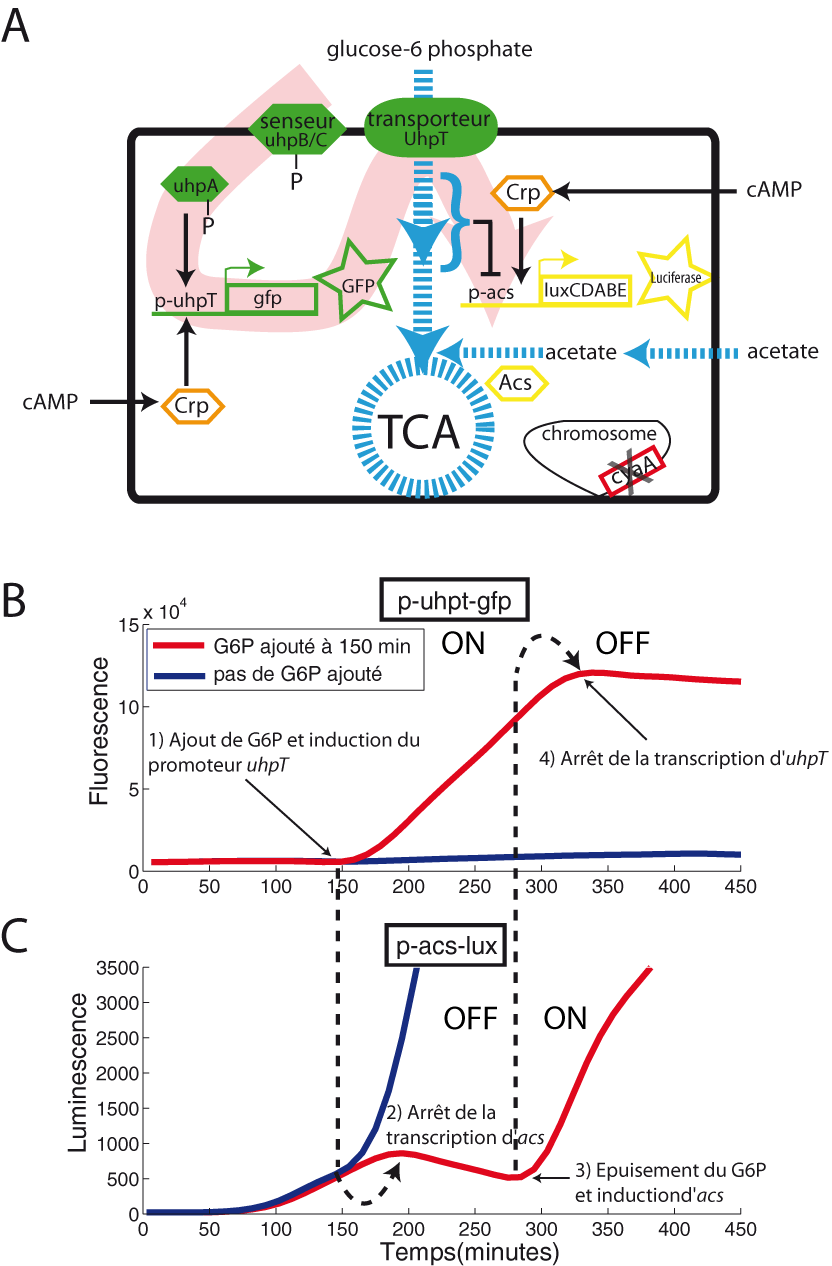
Système synthétique de communication intercellulaire par l'AMPc
Pour comprendre le mécanisme responsable de la répression catabolique, nous avons développé de nombreux outils (fusions transcriptionnelles, vecteurs de sur-expression inductibles, construction de souches mutantes). Cependant, ces efforts ne nous ont pas permis de percer à jour le mécanisme responsable de la répression catabolique. En échange, la nature nous a accordé la "serendipity" c'est-à-dire la découverte au hasard qui rend heureux.
En plaçant en contact un mutant Δcrp et un mutant ΔcyaA, nous nous sommes rendu compte que ces deux souches réussissaient à communiquer grâce à l'AMPc extracellulaire. L'expérience est relativement simple : nous avons placé une gélose contenant des bactéries Δcrp (dite "sender") en contact d'une gélose composée des bactéries ΔcyaA (dite "receiver") contenant un bio-senseur luminescent très sensible capable de détecter la présence d'AMPc extracellulaire. Ce biosenseur correspond au promoteur sdh (Crp-AMPc dépendant) fusionné à l'operon luxCDABE (plasmide p-sdh-lux). Après quelques heures, la gélose contenant les bactéries ΔcyaA s'est mise à émettre de la lumière. L'AMPc produit par la souche "sender" a diffusé dans la seconde gélose puis a pénétré dans les bactéries ΔcyaA, s'est fixé à Crp ce qui a permis l'activation du promoteur sdh puis l'émission de lumière (Figure 10A). Pour parfaire la démonstration, nous avons montré que la gélose "receiver" ne s'allume jamais si l'adenylate cyclase (production de l'AMPc) est absente dans la gélose "sender". De même, nous avons montré que la présence de Crp dans la gélose "receiver" est indispensable au processus de communication. Ces jeux avec le complexe Crp-AMPc nous ont permis d'illustrer "artistiquement", via la communication, le fonctionnement de cette jolie porte AND que constitue le complexe Crp-AMPc (Figure 10B).
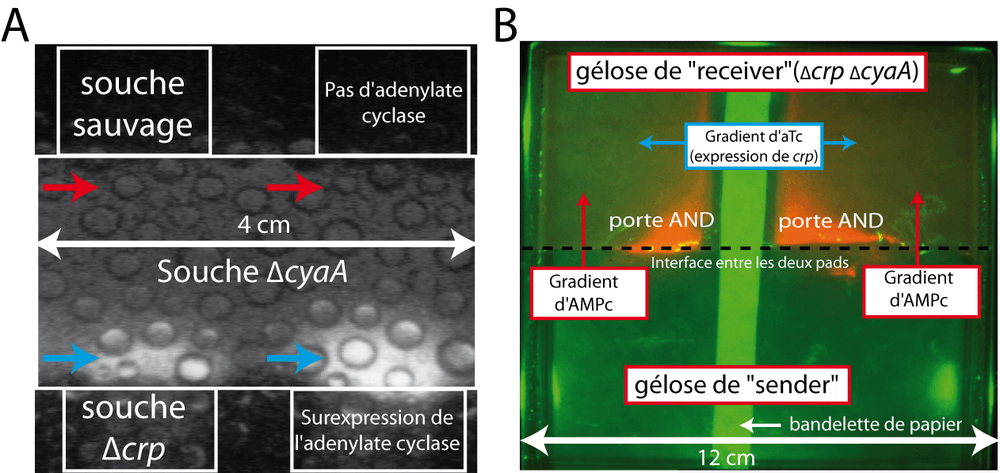
Pour caractériser plus précisément ce nouveau module de communication extracellulaire, nous avons également fait des expériences en milieu liquide. Ces expériences montrent que l'on peut contrôler très finement la communication en faisant varier la concentration d'adenylate cyclase dans les "senders " ou la concentration de Crp dans les "receivers". Un des avantages du complexe Crp-AMPc c'est qu'il est déjà connecté à de nombreuses autres fonctions cellulaires. Par exemple, un mutant ΔcyaA souffre d'un défaut de motilité lié à son incapacité à fabriquer des flagelles. Nous avons pu rétablir, par communication intercellulaire, la fabrication de flagelles et donc la motilité de bactéries ΔcyaA créant de fait une sorte de système de guidage synthétique. Cela montre qu'il est possible de connecter aisément notre système de communication à d'autres modules (physiologiques ou synthétiques) présents dans la cellule.
Ce processus de communication est-il physiologique ? L'AMPc secrété par la bactérie a-t-il pour fonction la communication? Nous ne sommes pas encore en mesure de répondre à cette question. Un résultat nous a particulièrement troublé : la limite basse de détection de l'AMPc extracellulaire de notre biosenseur (~5-10μM) correspond exactement à la limite haute de production d'AMPc extracellulaire par une souche sauvage (~1-10μM). C'est comme si les bactéries sauvages produisaient une concentration d'AMPc extracellulaire juste en dessous du seuil qui commencerait à modifier significativement l'activité du régulon Crp des autres bactéries. Nous n'avons pas d'explications à ce résultat surprenant.
Utiliser l'activité luciferase pour inférer l'état métabolique de la cellule
Le rapporteur luciferase a besoin de sources d'énergie cellulaires pour émettre des photons. Cette dépendance est souvent considérée comme un risque d'artefact majeur. Nous allons montrer qu'elle peut aussi être utilisée pour inférer de nombreuses informations sur l'état des cellules.
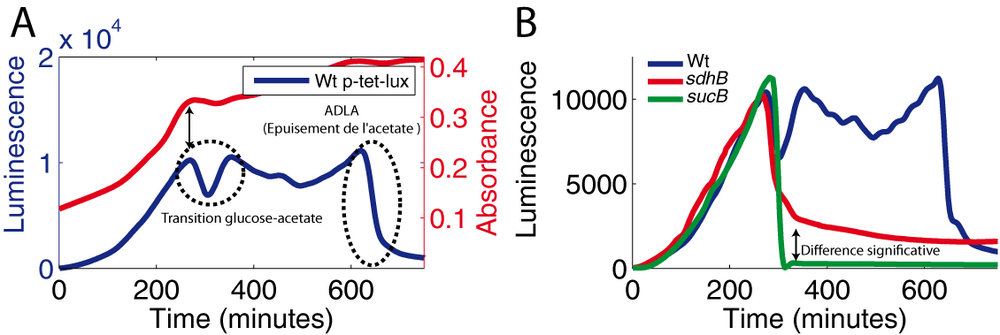
Pour émettre des photons, la luciferase que nous utilisons (Photorhabdus luminescens) ne nécessite pas l'ajout de substrat dans le milieu de culture. En effet, ce dernier est produit directement dans la cellule à l'aide de trois enzymes (encodées par luxC, luxD, luxE). La luciferase (encodée par luxA et luxB) oxydes simultanément ce substrat et le FMNH2 en présence d'oxygène pour émettre un photon à 490 nm. La production de FMNH2 requiert le fonctionnement des réactions centrales du métabolisme du carbone. Ainsi l'émission de lumière n'est pas seulement corrélée à la concentration de la luciferase mais également à l'activité du métabolisme central. Toute perturbation dans cette activité affecte la production de lumière. Cet effet peut être observé lorsque les bactéries épuisent la source de carbone de leur milieu et rentrent en phase stationnaire. A ce moment précis, on observe une chute de l'activité luciferase (que nous appellerons dorénavant ADLA, Abrupt Decline of Luciferase Activity) due à une chute du pouvoir réducteur (Koga et al., 2005). Il est facile de prouver que cette chute est liée à l'épuisement de la source d'énergie car l'ajout dans le milieu, après cet ADLA, de n'importe quelle source de carbone utilisable par les bactéries, rétablit quasi immédiatement l'émission de lumière (Figure 11).

Pour pouvoir étudier cette dépendance énergétique de la luciferase, nous nous sommes tout d'abord affranchi de toutes régulations transcriptionnelles. Nous avons fusionné un promoteur synthétique indépendant (tet) à l'operon luxCDABE (plasmide p-tet-lux). Avec cette construction, l'expression de la luciferase est sous notre contrôle : toutes les variations mesurées dans le signal de luminescence proviennent de changements d'états métaboliques. En faisant pousser des bactéries E. coli sauvages transformées avec ce senseur métabolique dans un milieu minimum contenant du glucose et de l'acétate, on observe deux phénomènes intéressants (figure 12A):
- il y a une légère diminution transitoire de luminescence lors de la transition glucose-acétate. Cet événement suggère une diminution transitoire du fonctionnement du métabolisme central.
- il y a une chute brutale de l'activité luciferase lorsque l'acétate est épuisé, c'est-à-dire lorsqu'il n'y a plus aucune source de carbone dans le milieu.
Cette méthode de mesure de l'état métabolique des cellules à l'aide d'un "biosenseur" est intéressante mais elle ne dévoile toute sa puissance que lorsqu'elle est couplée à une banque de simples mutants. Nous avons donc transformé des centaines de mutants avec notre biosenseur à la recherche de signaux qui diffèrent de la souche sauvage.
A titre d'exemple, le profil de luminescence d'un mutant sucB, déleté d'un gène codant pour une enzyme du cycle de Krebs, montre une chute brutale de l'activité luciferase non pas lors de l'épuisement de l'acétate (comme c'est le cas pour la souche sauvage) mais lors de l'épuisement du glucose (figure 12B). En effet, ce mutant ne peut pas utiliser l'acétate comme source de carbone car la néoglucogenèse est coupée. Une souche sdhB est aussi déletée d'un gène codant pour une enzyme du cycle de Krebs : on observe aussi une chute de l'activité luciferase lorsque le glucose est épuisé. Cependant l'ampleur de cette chute est bien moins prononcée que celle observée avec le mutant sucB. Il est très probable que cette différence de phénotype soit significative et mette en exergue l'existence d'une petite fuite métabolique permettant de maintenir un fonctionnement minimal du cycle de Krebs et donc une petite activité luciferase dans le mutant sdhB. Ainsi nous pensons que la capacité à maintenir l'activité luciferase associée à la forte sensibilité de détection de la lumière permettrait peut-être de détecter et de quantifier des variations fines dans les flux métaboliques. Menée de manière systématique (grand nombre de mutants), cette manière de procéder pourrait peut-être permettre de redessiner les flux métaboliques de manière plus quantitative dans une condition donnée.
La capacité à maintenir l'activité luciferase permet de se focaliser sur une enzyme, un flux spécifique. Ainsi nous avons pu mesurer, en temps réel, l'activité relative d'une enzyme : l'acétyl-coenzyme A synthétase encodée par le gène acs. L'idée consiste à créer et contrôler un goulot d'étranglement métabolique dans des bactéries saturées en luciferase. Nous avons donc supprimé toute trace d'acétyl-coenzyme A synthétase dans des bactéries placées dans un milieu minimum avec de l'acétate. Dans ces conditions, ces bactéries ne peuvent pas utiliser l'acétate : le métabolisme central est inactif. La luciferase, pourtant présente en forte concentration, ne peut pas émettre de photons. Puis nous rouvrons "la vanne" lentement: l'arrivée des premières enzymes d'acetyl-coenzyme A synthétase réamorce le métabolisme central qui peut alors utiliser l'acétate comme source d'énergie : la brusque remontée du pouvoir réducteur entraine le rétablissement de l'activité luciferase et le niveau de ce rétablissement est directement corrélé à la concentration de l'acetyl-coenzyme A synthétase. Avec cette méthode, il est donc possible de connaître son activité relative, directement in vivo et en temps réel.
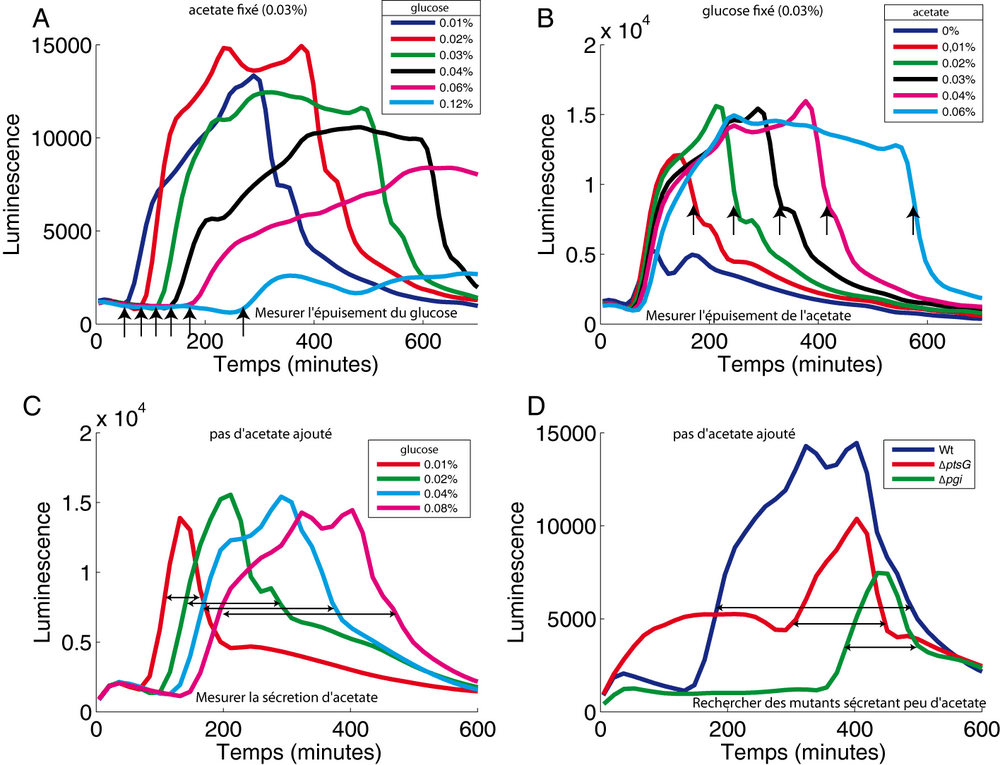
Enfin, l'utilisation du promoteur acs fusionné à l'operon luxCDABE (plasmide p-acs-lux) nous a permis d'inférer certains paramètres extérieurs, encore une fois directement et en temps réel. Nous avons vu que l'épuisement du glucose lève la répression exercée sur ce promoteur. Dans un milieu contenant du glucose et de l'acétate, l'induction de luminescence indique donc l'épuisement du glucose (Figure 13A). A l'inverse, la chute brutale de l'activité luciferase signale l'épuisement de l'acétate (Figure 13B). En associant ces deux événements, nous pensons qu'il est possible de mesurer la quantité relative d'acétate secrété par les bactéries dans le milieu c'est-à-dire le débordement du métabolisme (overflow metabolism). Lorsque les bactéries poussent sur glucose, le métabolisme central est saturé et une partie de l'énergie est évacuée vers l'extérieur sous forme d'acétate en vu d'une utilisation future. Pour mesurer ce phénomène, nous avons fait pousser des bactéries dans un milieu contenant uniquement du glucose. Nous avons remarqué que le temps entre l'induction de la luciferase (épuisement du glucose) et la chute de l'activité luciferase (épuisement de l'acétate) augmente proportionnellement avec la concentration de glucose utilisée (Figure 13C). L'explication la plus raisonnable pour expliquer l'accroissement de ce délai est l'augmentation de la concentration extérieure d'acétate. Pour étayer cette hypothèse, nous avons testé des mutants (ptsG, pgi) qui ralentissent la consommation du glucose (et donc la glycolyse) ce qui limite le débordement du métabolisme. Dans ces mutants et pour une même concentration de glucose, le délai entre l'induction de la luciferase et la chute de l'activité luciferase est beaucoup plus court comparé à une souche sauvage (Figure 13D). Ce résultat suggère, comme escompté, que la sécrétion d'acétate dans ces mutants est d'une ampleur beaucoup plus limitée.
Chapitres intermédiaires
Voir les publications en anglais qui décrivent plus précisément tous les résultats décrits ci-dessus.
- A genome-wide screen for identifying all regulators of a target gene
- Study of carbon catabolite repression in Escherichia coli by monitoring the activity of Crp-cAMP dependent promoters
- A synthetic Escherichia coli communication system mediated by extracellular cyclic AMP
- Using luciferase activity to infer the metabolic state of Escherichia coli
Conclusion et perspectives
La plus grosse partie expérimentale de ma thèse aura consisté à essayer de comprendre pourquoi l'ajout d'AMPc exogène ne déclenche pas l'expression du gène acs quand il y a encore du glucose dans le milieu. Pourtant, après avoir testé des centaines d'hypothèses et criblé l'ensemble des simples mutants d'E. coli, nous n'avons pas pu déterminer le mécanisme à l'origine de ce résultat. Notre apport a donc principalement consisté à déconstruire les deux modèles principaux expliquant le phénomène de répression catabolique. Nous avons ainsi montré que la variation intracellulaire d' [AMPc] ne peut pas être à l'origine de la répression catabolique. Ce constat est d'autant plus paradoxal que l'AMPc joue neamoins un rôle de premier plan dans le processus. De même, nous avons montré que l'état de phosphorylation du PTS ne peut pas expliquer la répression catabolique malgré le fait que, encore une fois, le PTS y joue indéniablement un rôle. Nous proposons, humblement et sans certitude, de recentrer les recherches sur la protéine Crp et les variations de flux glycolytique (indépendamment du ratio PEP/pyruvate): ce sont les deux seuls éléments qui, dans nos conditions, se sont avérés indispensables au fonctionnement de l'interrupteur. Une hypothèse potentielle proposée par Magasanik il y a maintenant 50 ans (Magasanik, 1961) est celle du métabolite répresseur : peut-on imaginer un deuxième petit métabolite qui contrôlerait l'expression de Crp sans que nous nous en soyons rendu compte jusqu'à maintenant ? La solution réside-elle dans un mécanisme plus complexe, encore inconnu, et difficile à détecter avec les méthodes actuelles? Ou, la solution nous est-elle tout simplement passée sous les yeux ? L'avenir nous le dira.
Ainsi, c'est sur ces recherches restées bloquées dans une impasse que se sont greffés les résultats qui suivent. Ces derniers sont plus sûrs, plus facilement reproductibles, plus intéressants et débouchent a priori sur bien plus d'applications. Ils n'ont cependant nécessité qu'une petite fraction de mon temps de travail expérimental.
L'étude de l'opéron araBAD est un cas d'école pour décrire les variations stochastiques dans l'expression des gènes. Dans nos expériences, nous avons pu montrer qu'un flux glycolytique fort a tendance à synchroniser l'expression de l'opéron araBAD alors que la diminution de ce flux à l'aide d'un sucre non métabolisable module la distribution d'expression de cet operon dans la population de cellules. Les modélisateurs pourraient tenter de comprendre ce phénomène et inclure le flux glycolytique dans leur modèle stochastique.
Notre interrupteur métabolique, basé sur les activités inversées des promoteurs acs et uhpT en fonction du flux glycolytique, est très robuste, facile à utiliser et très rapide : l'inversion des expressions est visible en une dizaine de minutes. Il possède cependant deux défauts majeurs : on ne sait pas comment il fonctionne, car il est basé sur le mécanisme inconnu de la répression catabolique. De plus, il manque d'orthogonalité c'est-à-dire qu'il n'est pas du tout indépendant/découplé du réseau de régulation de la bactérie. Nous espérons cependant que notre système séduira des biologistes synthétiques ou des modélisateurs.
Un autre système synthétique développé dans cette thèse est le système de communication intercellulaire par l'AMPc. Ce système est robuste et facilement modulable, mais il souffre également du manque d'orthogonalité : il est directement connecté au "hub" que représente le complexe Crp-AMPc. Or, ce défaut a priori peut être transformé en avantage car ce complexe, très documenté dans la littérature, est déjà connecté à de nombreux modules (motilité, chimiotactisme, métabolisme, virulence, résistance au stress) et compose un grand nombre de motifs récurrents comme les boucles de type "feed forward". Il existe, en outre, un très grand nombre de promoteurs contrôlés par le complexe Crp-AMPc et donc un large choix d'affinité de sites de liaison. Celui qui voudrait s'emparer de la difficile question de "la modularité" des réseaux de régulation génique pourrait donc trouver ici un terreau expérimental favorable.
Dans notre système de communication synthétique, il a fallu construire et optimiser deux souches. Une usine microbienne qui fabrique de fortes concentrations d'AMPc extracellulaire et un bio-senseur qui en détecte de très faibles. L'usine bactérienne est capable de produire plus de 150μM d'AMPc dans le milieu extérieur soit ~10-100 fois plus qu'une souche sauvage. Cette concentration reste très éloignée des 30mM produit par Microbacterium sp. no. 205. Il est donc peu probable que notre souche soit compétitive à l'échelle industrielle. Cependant, ce rendement pourrait être amélioré en supprimant la phosphodiesterase (le gène cpdA) qui a justement un Km de l'ordre de 500μM. Il n'est donc pas totalement exclu que l'amélioration de cette souche la rende compétitive. Le bio-senseur, lui, a sa limite basse de détection de l'AMPc extracellulaire vers 5-10μM. Or le Km de l'AMPc pour Crp est dix fois plus faible. On sait que l'export de l'AMPc est actif, mais le gène impliqué dans cet export n'a pas encore été découvert. Pour espérer "récupérer" ce facteur 10, il faudrait donc déterminer quels sont les gènes impliqués dans l'import / export de l'AMPc, puis il faudrait bloquer l'export et facilité l'import.
Il reste également à déterminer si le processus de communication intercellulaire par l'AMPc n'existe que dans les mains du biologiste synthétique ou s'il s'agit d'un mécanisme physiologique. Pourquoi la cellule gaspillerait-elle 9% de ses réserves en ATP à secréter de l'AMPc (Matin & Matin, 1982) ? Ce processus de communication, s'il il existe, n'est pas forcement "inter-bactérien" : il pourrait potentiellement être "inter-royaume", car l'AMPc est une molécule de signalisation utilisée également par les cellules eucaryotes. Nous avons entrepris des expériences visant à montrer un processus de communication entre nos cellules et l'amibe sociale Dictyostelium discoideum, sans succès néanmoins, faute de temps (Figure 14). Cependant, c'est aussi l'énorme avantage de notre petit système synthétique : il peut être implémenté entre des royaumes différents. Nous espérons donc que d'autres réussiront là où nous avons dû renoncer.

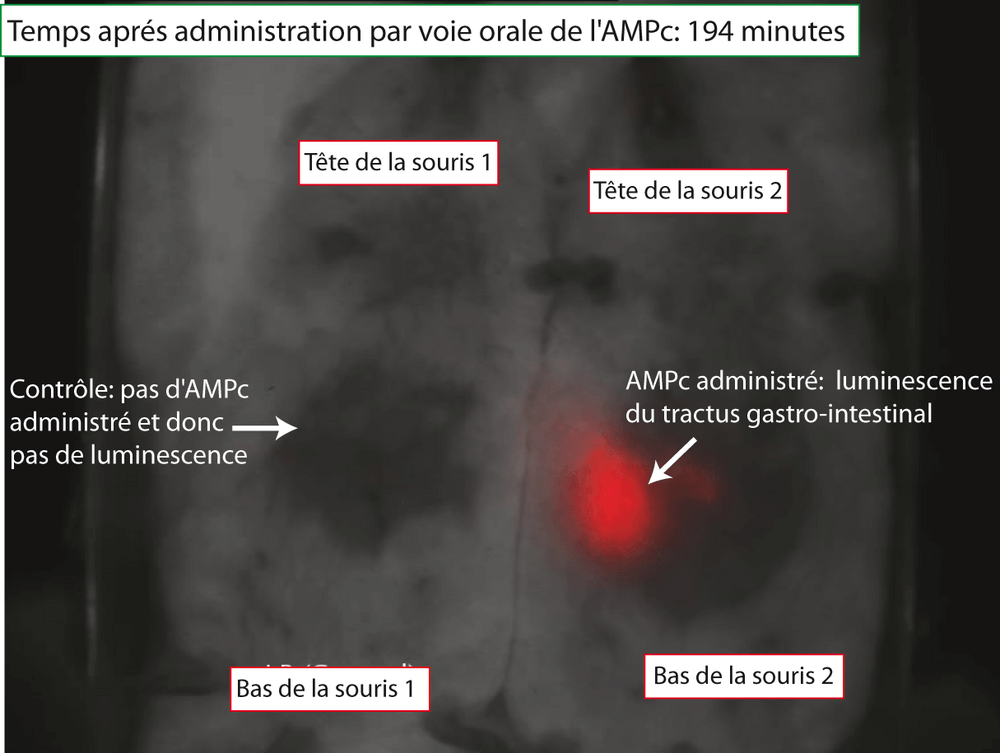
Enfin, le milieu le plus logique où pourrait prendre place cette communication via l'AMPc chez Escherichia coli est l'écosystème gastro-intestinal. Les conditions y sont très différentes des conditions de laboratoire et la densité de cellules beaucoup plus forte pourrait faciliter un processus physiologique de communication. Pour tester cette hypothèse, nous avons inoculé à des souris, par voie orale, la souche bio-senseur. L'idée était que le bio-senseur s'allumerait dans les intestins de la souris grâce à un processus de communication avec une souche "sender" physiologique. L'expérience n'a pas pu démontrer de processus de communication. Cependant, nous avons ensuite administré, par voie orale, de l'AMPc à la souris. Puis, nous avons observé un accroissement de lumière dans la zone du tractus gastro-intestinal et ce, de manière non-invasive, c'est-à-dire transcutané (Figure 15). Dans cette expérience, nous avons donc induit l'expression d'un gène (l'opéron luxCDABE) de bactéries placées dans le tractus gastro-intestinal d'une souris et nous avons contrôlé visuellement la réussite de l'induction. Ce type d'expériences pointe du doigt des technologies plus futuristes où nos intestins seront inspectées par des cellules médicaments, véritables micro-ordinateurs synthétiques, chargées de surveiller et de rétablir l'homéostasie de l'hôte ou de signaler la présence d'un pathogène.
Dans ces expériences de communication, il perdure une sorte de regret "artistique" : malgré des dizaines de tentatives, nous n'avons jamais pu filmer le processus de communication par l'AMPc, sous le microscope, à l'échelle de la cellule. Nous attendons donc que quelqu'un relève ce défi et nous envoie les vidéos. [2023: L’expérience a été réussi quelques mois plus tard et la vidéo est fourni dans les informations supplémentaires de la publication correspondante.]
Il reste maintenant à décrire les perspectives liées à l'utilisation de l'activité luciferase pour inférer de nombreux paramètres in vivo et en temps réel sur le fonctionnement de la cellule. Il s'agit, sans doute, des résultats qui auront le plus d'impact, même si paradoxalement ce sont aussi ceux qui ont été les plus faciles à obtenir. La combinaison de la mesure de l'activité luciferase, des banques de mutants et du savoir actuel sur le métabolisme pourrait aider à générer des données quantitatives plus précises sur l'état des flux métaboliques dans une condition donnée. L'avantage de cette technique est qu'elle ne nécessite pas d'étapes d'échantillonnage et de "quenching" comme c'est le cas pour les mesures de metabolome effectuées a posteriori de l'éclatement des cellules.
La capacité à maintenir l'activité luciferase apporte de nombreuses informations in vivo et en temps réel : citons l'état énergétique global, l'état du regulon Crp, l'état de la machinerie globale, l'état du milieu, mais aussi des informations plus précises sur l'état d'un flux ou d'une enzyme particulière. La facilité, la sensibilité et le faible coût de détection de la lumière rend notre méthode relativement facile à implémenter. Elle pourrait avoir de nombreuses applications en biotechnologie, en biologie des systèmes ou en biologie synthétique. L'utilisation de notre méthode en fermenteur industriel semble possible a priori : il n'est même pas nécessaire que toutes les cellules disposent de la luciferase (ce qui risquerait d'avoir un coût énergétique trop élevé) : une petite proportion de cellules, présentes dans le fermenteur et saturées en luciferase, collecteraient les informations nécessaires, informations elles-mêmes récupérées en temps réel par un simple photomultiplicateur.
A l'opposé, et même si cela nécessite sans doute un matériel plus coûteux, il n'est pas impossible que notre méthode soit implémentable à l'échelle de la cellule isolée. En effet, des cameras sensibles, placées sur un microscope, arrivent à détecter la luminescence de cellule unique (Mihalcescu et al., 2004). Nous pourrions alors suivre, en temps réel, l'état du métabolisme central de cellules isolées ainsi que tous les autres paramètres cités précédemment.
Enfin nous terminerons par vanter les mérites de la fusion transcriptionnelle p-acs-lux rapportant l'activité du promoteur acs grâce à la luciferase. Il s'agit d'un bio-senseur robuste et facile à utiliser. Placé dans un milieu minimum avec du glucose et de l'acétate, il rapporte l'épuisement du glucose (induction de la luciferase) et l'épuisement de l'acétate (chute brutale de la luminescence). Placé dans un milieu minimum avec du glucose uniquement, il rapporte la concentration d'acétate sécrétée dans le milieu extérieur c'est-à-dire le débordement du métabolisme (overflow metabolism). L'accumulation de l'acétate dans le milieu de culture est un problème important dans la fermentation industrielle car cet acide organique inhibe la croissance cellulaire et donc la production des protéines recombinantes. Différentes approches sont actuellement développées pour tenter de réduire l'accumulation d'acétate en modifiant les flux du métabolisme central (Gosset, 2005). Notre bio-senseur pourrait potentiellement aider à mesurer, en temps réel, l'acétate secrété voire même à cribler des banques de mutants à la recherche de candidats intéressants secrétant peu d'acétate.
Bibliographie
- Crasnier-Mednansky, M. 2008. Is there any role for cAMP-CRP in carbon catabolite repression of the Escherichia coli lac operon ? Nat Rev Microbiol, 6(12), 954 ; author reply 954. 13
- Gorke, B., & Stulke, J. 2008. Is there any role for cAMP-CRP in carbon catabolite repression of the Escherichia coli lac operon ? Reply from Gorke and Stulke. Nat Rev Microbiol, 6(12), 954. 13
- Gosset, G. 2005. Improvement of Escherichia coli production strains by modification of the phosphoenolpyruvate :sugar phosphotransferase system. Microb Cell Fact, 4(1), 14. 132 Magasanik, B. 1961. Catabolite repression. Cold Spring Harb Symp Quant Biol, 26, 249–56. 127
- Inada, T., Kimata, K., & Aiba, H. 1996. Mechanism responsible for glucose-lactose diauxie in Escherichia coli : challenge to the cAMP model. Genes Cells, 1(3), 293–301. 13
- Keseler, I. M., Collado-Vides, J., Santos-Zavaleta, A., Peralta-Gil, M., Gama-Castro, S., Muniz- Rascado, L., Bonavides-Martinez, C., Paley, S., Krummenacker, M., Altman, T., Kaipa, P., Spaulding, A., Pacheco, J., Latendresse, M., Fulcher, C., Sarker, M., Shearer, A. G., Mackie, A., Paulsen, I., Gunsalus, R. P., & Karp, P. D. 2010. EcoCyc : a comprehensive database of Escherichia coli biology. Nucleic Acids Res, 39(Database issue), D583–90. 1
- Khankal, R., Chin, J. W., Ghosh, D., & Cirino, P. C. 2009. Transcriptional effects of CRP* expression in Escherichia coli. J Biol Eng, 3(1), 13. 8 Koga, K., Harada, T., Shimizu, H., & Tanaka, K. 2005. Bacterial luciferase activity and the intracellular redox pool in Escherichia coli. Mol Genet Genomics, 274(2), 180–8. 17
- Kumari, S., Tishel, R., Eisenbach, M., & Wolfe, A. J. 1995. Cloning, characterization, and functional expression of acs, the gene which encodes acetyl coenzyme A synthetase in Escherichia coli. J Bacteriol, 177(10), 2878–86. 2
- Kumari, S., Beatty, C. M., Browning, D. F., Busby, S. J., Simel, E. J., Hovel-Miner, G., &Wolfe, A. J. 2000. Regulation of acetyl coenzyme A synthetase in Escherichia coli. J Bacteriol, 182(15), 4173–9. 2
- Matin, A., & Matin, M. K. 1982. Cellular levels, excretion, and synthesis rates of cyclic AMP
- in Escherichia coli grown in continuous culture. J Bacteriol, 149(3), 801–7. 129
- Megerle, J. A., Fritz, G., Gerland, U., Jung, K., & Radler, J. O. 2008. Timing and dynamics of single cell gene expression in the arabinose utilization system. Biophys J, 95(4), 2103–15 12
- Meighen, E. A. 1991. Molecular biology of bacterial bioluminescence. Microbiol Rev, 55(1) 123–42. 6
- Mihalcescu, I., Hsing, W., & Leibler, S. 2004. Resilient circadian oscillator revealed in individual cyanobacteria. Nature, 430(6995), 81–5. 131
- Narang, A. 2009. cAMP does not have an important role in carbon catabolite repression of the Escherichia coli lac operon. Nat Rev Microbiol, 7(3), 250. 13
- Oh, M. K., Rohlin, L., Kao, K. C., & Liao, J. C. 2002. Global expression profiling of acetategrown Escherichia coli. J Biol Chem, 277(15), 13175–83. 2
- Saier, M. H., & Roseman, S. 1972. Inducer exclusion and repression of enzyme synthesis in
- mutants of Salmonella typhimurium defective in enzyme I of the phosphoenolpyruvate : sugar phosphotransferase system. J Biol Chem, 247(3), 972–5. 8
- Veening, J. W., Smits, W. K., & Kuipers, O. P. 2008. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol, 62, 193–210. 2
- Wolfe, A. J. 2005. The acetate switch. Microbiol Mol Biol Rev, 69(1), 12–50. 2
- Zaslaver, A., Bren, A., Ronen, M., Itzkovitz, S., Kikoin, I., Shavit, S., Liebermeister, W., Surette, M. G., & Alon, U. 2006. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods, 3(8), 623–8. 10